
Reduction of ferrylmyoglobin by cysteine as affected by pH†
Abstract
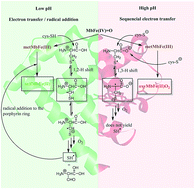
Reduction of the hypervalent meat pigment ferrylmyoglobin, MbFe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, by cysteine is enhanced by acid due to protonation of ferrylmyoglobin to yield sulfmyoglobin as the main product, while at alkaline conditions, the rate decreases with the cysteine dianion as the reactant forming oxymyoglobin, MbFe(II)O2. The second-order rate constant for cysteine reacting with protonated ferrylmyoglobin is 5.1 ± 0.4 L mol−1 s−1 at 25 °C in 0.16 M aqueous sodium chloride and for the cysteine dianion reacting with ferrylmyoglobin 0.31 ± 0.15 L mol−1 s−1. For pH = 7.4 the activation parameters for sulfmyoglobin formation is ΔH‡ = 75 ± 2 kJ mol−1 and ΔS‡ = −250 ± 7 J mol−1 K−1 with similar values for homocysteine and glutathione. The difference in product is indicative of a shift from an electron-transfer/radical addition mechanism at low pH as in the stomach to a two-step electron-transfer mechanism at higher pH as in the intestine, and is discussed in relation to protection against the formation of radicals by sulphurous compounds during the digestion of red meat.
O, by cysteine is enhanced by acid due to protonation of ferrylmyoglobin to yield sulfmyoglobin as the main product, while at alkaline conditions, the rate decreases with the cysteine dianion as the reactant forming oxymyoglobin, MbFe(II)O2. The second-order rate constant for cysteine reacting with protonated ferrylmyoglobin is 5.1 ± 0.4 L mol−1 s−1 at 25 °C in 0.16 M aqueous sodium chloride and for the cysteine dianion reacting with ferrylmyoglobin 0.31 ± 0.15 L mol−1 s−1. For pH = 7.4 the activation parameters for sulfmyoglobin formation is ΔH‡ = 75 ± 2 kJ mol−1 and ΔS‡ = −250 ± 7 J mol−1 K−1 with similar values for homocysteine and glutathione. The difference in product is indicative of a shift from an electron-transfer/radical addition mechanism at low pH as in the stomach to a two-step electron-transfer mechanism at higher pH as in the intestine, and is discussed in relation to protection against the formation of radicals by sulphurous compounds during the digestion of red meat.
 Please wait while we load your content...
Please wait while we load your content...

