
Ultrastructure of metallopeptide-based soft spherical morphologies†
Abstract
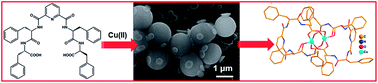
Peptides and proteins offer interesting starting points for triggering self-assembly processes owing to the chemical diversity of side-chains, ease of chemical modifications and the possibility of exploiting several non-covalent and metal-assisted interactions, to stabilize higher order ensembles. Consequently, a variety of nanoscale morphologies such as fibers, vesicles, nanotubes are observed for modified amino acids and short peptides and these biocompatible soft materials have been used for diverse biological, medical and material applications. Herein, we report metal-mediated modification of spherical soft assemblies, by introducing a coordinating linker for the Phe–Phe dipeptide, which results in the coalescence of soft structures. The possibility of copper ion coordination, with the metal-binding peptide conjugate, was confirmed by single crystal analysis. Based on these observations, a model depicting possible interactions leading to soft structure formation and metal-aided coalescence is also presented. The coalescence could be reversed in the case of Au-mediated soft structures with the help of thiol interference. Such an approach, exploiting interfacial metal ion interactions, is expected to provide an entry into novel metallopeptide materials.
 Please wait while we load your content...
Please wait while we load your content...

