
Photofragment ionization-loss stimulated Raman spectroscopy of a hydrated neurotransmitter: 2-phenylethylamine–water†
Nitzan Mayorkas,
Snir Cohen,
Hanan Sachs and
Ilana Bar*
Department of Physics, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel. E-mail: ibar@bgu.ac.il
First published on 24th October 2014
Abstract
Different types of spectral signatures and their interpretation provide valuable information on intra- and inter-molecular interactions that lead to unique shapes of molecules and clusters. Here, ionization-loss stimulated Raman (ILSR) spectra of the 2-phenylethylamine–water (PEA–H2O) cluster were obtained by monitoring the spectral signatures of PEA and of CH2NH2–H2O (a hydrated portion of the PEA tail) fragment ions. The ILSR spectra of both fragments were found to be similar, indicating that they are actually reminiscent of the PEA–H2O cluster signature. Comparison of the photofragment spectra to anharmonic calculated Raman spectra of the different conformers of the PEA–H2O clusters, suggests a structure with PEA similar to that of the most stable gauche conformer, with H2O hydrogen bonded to the nitrogen lone pair.
1 Introduction
The investigation of the spatial arrangements of biomolecules and of the forces that lead to them is usually adopted as a reductionist approach1 towards eventual understanding of molecular processes playing a central role in biospecific recognition. In principle, the structures of isolated molecules are a result of intra-molecular interactions and in other environments a consequence of a delicate balance between intra- and inter-molecular interactions. Therefore, obtaining information regarding these interactions is of particular importance for getting insight into the structures and intrinsic molecular properties of isolated biomolecular building blocks and their clusters.2–9Structures are usually revealed by comparing the spectroscopic results for molecules or their clusters in the gas phase, to high level quantum calculations. Indeed, a variety of experimental techniques, including electronic, vibrational and rotational spectroscopies combined with results of quantum calculations are employed to assess detailed structural pictures of the conformational landscapes of biomolecules.
A molecule that attracted considerable interest is the 2-phenylethylamine (C6H5–CH2–CH2–NH2, PEA) that contains a rigid skeleton ring and a flexible ethylamino side chain.10–18 This model system reflects the parent structure of a group of amine compounds functioning in biological systems as neurotransmitters and neuromodulators and was observed to fold into several different conformers in the gas phase. Different methods revealed the presence of four,10 two,16 and five conformers,11 where the fifth one13 was reassigned to a water cluster. Eventually, the presence of four conformers was confirmed by additional experimental investigations combined with the results of quantum calculations.12,14,15,17,18
These studies revealed that the flexibility of the ethylamine side chain in PEA, which is a consequence of the two principal flexible coordinates, including internal rotation about the Cα–Cβ and Cα–N single bonds, affords the existence of the conformers. In particular it has been found that the two lowest and two highest energy conformers of PEA correspond to structures with the ethylamino side chain folded (gauche) and extended (anti). In the gauche conformers, the amino hydrogen atoms are oriented differently toward the aromatic ring to form unique weakly N–H⋯π hydrogen bonded (HB) structures, slightly stabilizing the isolated molecules. On the other hand, in the anti conformers, the amino hydrogen atoms with symmetric and nonsymmetric orientations, point far away from the ring, leading to less stable conformers, due to HB absence.
The PEA–water clusters were only scantily studied, using laser induced fluorescence excitation and one- and two-color mass-selected resonant two-photon ionization (R2PI) of the S1 ← S0 electronic transition, the M(H2O)n (n = 1–4) clusters were detected.13,14 Their stoichiometry was assigned on the basis of the observed ion fragmentation patterns, band contour analysis and calculated energy differences between the conformers. Very recently,19 the rotational spectrum of the most stable PEA–H2O molecular adduct was studied by free jet absorption microwave spectroscopy.
Nevertheless, vibrational spectral signatures of the PEA–H2O clusters were not measured yet. In the present work ionization-loss stimulated Raman spectroscopy (ILSRS),18,20–22 where the depletion of ground state species by SR excitation is probed by R2PI, was used. However, in contrast to our previous studies that measured the spectra of different parent ions, here we were interested in testing whether it would be possible to reveal the structure of the PEA–H2O cluster by photofragment ILSR spectroscopy. Hence, the ILSR spectra of the fragment ions, i.e., PEA and CH2NH2–H2O (the hydrated portion of the PEA tail) were measured, enabling to infer the characteristic vibrational modes of the PEA–H2O parent cluster in an extended spectral range. The acquired spectra were compared to the calculated anharmonic Raman spectra of the PEA–H2O cluster, obtained by density functional theory (DFT) calculations. The structure of the PEA–H2O cluster could be determined by choosing the calculated spectrum that showed the best overall agreement with the measured photofragment ILSR spectra.
2 Methods
2.1 Experimental
A pumped Wiley–McLaren home built time-of-flight mass spectrometer (TOFMS), resembling the design of ref. 23, was used. The TOFMS consists of differentially pumped source and main detection chambers, separated by a skimmer with a 2.0 mm diameter (dia.) aperture. PEA–H2O clusters were formed by passing a stream of argon, saturated with water, at a pressure of about 1 bar, through the vapor of a PEA sample, purchased from Sigma Aldrich (99% pure), held in a reservoir at room temperature. The gas mixture was then expanded through a pulsed nozzle valve (General Valve, Series 9, 0.8 mm orifice) operating at 10 Hz and then through the skimmer before entrance into the main vacuum chamber. The generated molecular beam was introduced into the main chamber, while pointing toward the detection plane and passing through the hole of the repeller.In the interaction region of the TOFMS, the molecular beam was intersected by the laser beams, allowing measurement of mass-selected R2PI and ILSR spectra. R2PI spectra were measured using a tunable frequency-doubled pulsed neodymium:yttrium aluminum–garnet (Nd:YAG)-pumped dye laser (UV) with ∼5 ns pulses at a 10 Hz frequency. For monitoring ILSR spectra, an additional Nd:YAG laser system was employed to provide the SRS beams, while parking the UV laser on resonances, corresponding to the particular species. The additional laser provided the second harmonic (532 nm) with ∼5 ns pulses at 10 Hz, which was split in a one to four ratio, so that the former supplied the pump beam, ωp, and the latter pumped a dye laser to generate a tunable ωS beam. The vertically polarized ωp and ωS beams, with energies of ∼17 mJ and 25 mJ, respectively, were counterpropagated and used to induce the Raman transitions. The ωp and ωS beams were focused with 35 and 30 cm focal length (f.l.) planoconvex lenses and aligned to spatially and temporally overlap in the interaction region of the TOFMS. These beams also overlapped the UV beam, focused by the 35 cm f.l. lens, and preceded the UV beam by ∼30 ns.
Ions obtained via R2PI and ILSR were detected by a micro-channel plate (MCP) and fed into a fast preamplifier, allowing measurement of mass spectra with a fast digital oscilloscope and recording of the integrated intensity of the ion peak of interest. The spectra were monitored by measuring the integrated ion signal as a function of the UV or ωS beam wavelengths. For each of the species, three to eight spectra were measured and averaged to achieve suitable signal-to-noise levels.
2.2 Calculations
Quantum mechanical DFT calculations and data treatment were performed, according to our approach in ref. 22, to aid in interpreting the measured ILSR spectra. The calculated spectra were obtained following initial geometry optimizations of the conformers of PEA molecules and of PEA–H2O clusters, using the MMFF94s force field in Avogadro.24 Then, these initial geometries were introduced in the GAUSSIAN 09 package25 and optimized using “tight” self-consistent field convergence criteria and “ultrafine” integration grids. The optimization of the geometries of the conformers was performed by the Becke three parameter hybrid functional combined with Lee–Yang–Parr correlation functional (B3LYP)26,27 and the 6-311++G(d,p) basis set. These geometries were used for calculation of harmonic and anharmonic vibrational frequencies and of Raman activities at the same level of theory. In addition, single-point energy calculations for finding minimum energies were carried out by second order Møller–Plesset theory (MP2)28 with the same basis set.2.3 Determination of the calculated spectrum that best fits the measured one
The Euclidean distances related to the frequencies and intensities of the spectra were evaluated, while manually choosing the peaks from the experimental and theoretical spectra. These sets were then used to construct four new vectors of wavenumbers and intensities, i.e., Vexp, Iexp, Vtheo and Itheo, where Vexp,theo were the measured and calculated peak wavenumbers and Iexp,theo – the respective peak intensities. The experimental and theoretical peaks were then arranged in pairs according to the fitting rule of minimal Euclidian distance: , where ωexp,theo was the distance between an experimental peak and its matched calculated peak. The average of the distances for each correlated peak pair gave a fitting quality indicator.
, where ωexp,theo was the distance between an experimental peak and its matched calculated peak. The average of the distances for each correlated peak pair gave a fitting quality indicator.
3 Results and discussion
3.1 Resonant two-photon ionization
Mass-selected one-color R2PI spectra of PEA (m/z = 121), CH2NH2–H2O (m/z = 48) and of PEA–H2O (m/z = 139) in the region of the S1 ← S0 transitions are shown in Fig. 1 panels (a), (b) and (c), respectively. It is clearly seen that the PEA spectrum shows the A–E dominant features, labeled according to ref. 13 and 14, and the other spectra show only single features. Actually, the A–D features were attributed to the four conformers of PEA,10,13–15,18 while peak E was suggested to be a result of PEA–H2O cluster fragmentation.13,14 This is supported by the appearance of a nearly coincident peak in the fragmentation channel, m/z = 48, corresponding to the hydrated portion of the ethylamino tail and resulting from Cα–Cβ bond cleavage. As already suggested,14 the observation of this fragment can be considered as an indication that the PEA–H2O cluster consists of a water molecule fused to the terminal amino group of PEA. Observation of the fragments related to only one of the conformers of the PEA–H2O cluster is in line with the results of Melandri et al.19 When they used Ar as carrier gas, or even He, they could not find rotational transitions belonging to other conformers that could have relaxed in the jet. Thus the addition of a single water molecule to PEA is considered to be accompanied by the collapse of its four conformers into a single structure, with the ethylamino side chain in a conformation related to the most highly populated conformer of the PEA monomer (see below). | ||
| Fig. 1 One-color mass-selected resonantly enhanced two photon ionization spectra of (a) 2-phenylethylamine (PEA) (m/z = 121), and of (b) CH2NH2–H2O (m/z = 48) and (c) PEA–H2O (m/z = 139) fragments in the S1 ← S0 origin region. The vertical dashed line indicates the position of the feature related to PEA and CH2NH2–H2O fragments. | ||
Furthermore, examination of the R2PI spectrum, corresponding to the mass channel of PEA–H2O, Fig. 1(c), shows only a feature shifted to the red (relative to E), but none of the A–E bands. Hence, this feature is attributed to a photofragment, implying that ionization of PEA water clusters does not afford observation of the parent ions. This agrees with previous results, which pointed out that any of the PEA–water clusters, even with near-threshold ionization lose at least one water molecule13,14 and this feature was identified to be a fragment resulting from the PEA–(H2O)3 parent.
This behavior, where only fragments resulting from the parent clusters could be observed in the R2PI spectra may be explained by the dynamics in the ionic states. For example, feature E, comes from the excitation of a PEA–H2O cluster and not from ionization of the PEA monomer. Nevertheless, its intensity is comparable to peaks A–D [Fig. 1(a)] that are associated with the PEA monomers. Since normally the number of clusters is much smaller than the number of monomers in the molecular beam, the transition probability for excitation of the cluster itself must be large to account for the large signal. By assuming that the electronic factors in the transition probability are the same for PEA and PEA–H2O, it follows that the Franck–Condon (FC) factors for the cluster must not be smaller than the FC factors for PEA, thus leading to preparation of the excited (ionic) state that evolves to fragments, preventing the observation of the parent ion.
3.2 Ionization-loss stimulated Raman spectra
Although we were not able to directly monitor the R2PI spectrum of the PEA–H2O parent ion, we were interested in testing whether it would be possible to obtain information on the PEA–H2O cluster by monitoring ILSR spectra of the photofragments. Therefore, the ILSR spectra of the most stable PEA monomer and of the PEA and CH2NH2–H2O fragments were measured, while parking the exciting UV laser on the C and E bands, Fig. 1(a), and on the band in Fig. 1(b), respectively. Fig. 2(a) shows the ILSR measured spectrum of the PEA fragment, and at the top of Fig. 3(a) and (b) and in Fig. 3(c), the ILSR measured spectra of the PEA monomer and of the PEA and CH2NH2–H2O fragments, respectively, appear. These measured spectra, consist of sharp Q-branches of the transitions to different vibrational modes. Fig. 2 also includes the calculated Raman spectra, the electronic ground state, S0, structures, as well as the zero-point energy corrected electronic energies, evaluated by MP2/6-311++G(d,p) single point calculations with zero-point vibrational energy corrections from B3LYP/6-311++G(d,p)] levels of theory, of the gauche and anti conformers of the PEA–H2O clusters, i.e., PEA(GI)-W, PEA(GIII)-W, PEA(GII)-W, PEA(AI)-W and PEA(AII)-W,19 in panels (b)–(f), respectively. The calculated Raman spectra are based on anharmonic vibrational frequencies and Raman intensities at the B3LYP/6-311++G(d,p) level of theory and they correspond to the electronic ground state, S0, structures of the conformers of PEA–H2O.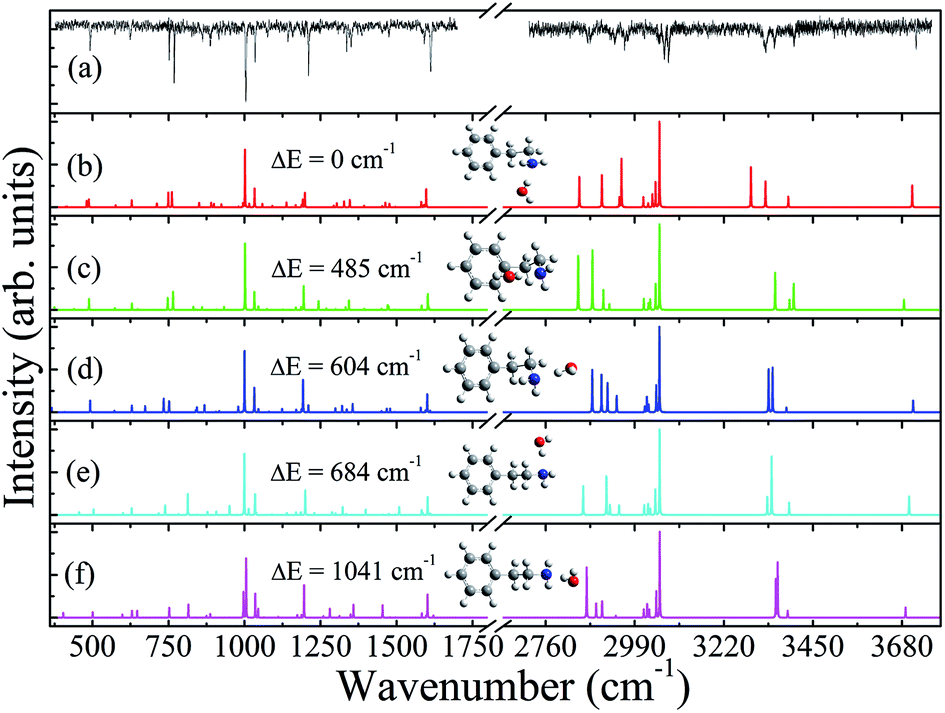 | ||
| Fig. 2 (a) Ionization-loss stimulated Raman spectrum, obtained when the UV laser is parked on the feature E related to the 2-phenylethylamine (PEA) fragment in Fig. 1(a) and panels (b)–(f) represent the theoretical spectra of the PEA–H2O clusters, corresponding to the shown electronic ground state, S0, structures of PEA(GI)-W, PEA(GIII)-W, PEA(GII)-W, PEA(AI)-W and PEA(AII)-W, respectively. The calculated spectra are based on calculated anharmonic vibrational frequencies and Raman intensities at the B3LYP level with a 6-311++G(d,p) basis set for the corresponding structures and convoluted with Lorentzian lines of full width half maximum of 0.5 cm−1. In each panel the geometries and the zero-point energy corrected electronic energies of the PEA-W conformers, as evaluated by single point Møller–Plesset calculations with the 6-311++G(d,p) basis set are shown. | ||
 | ||
| Fig. 3 Ionization-loss stimulated Raman spectra obtained when the UV laser is parked on the features C (a) and E (b) related to 2-phenylethylamine (PEA) in Fig. 1(a) and on the hydrated tail portion (c) in Fig. 1(b). At the bottom of panels (a) and (b) the theoretical spectra of PEA and of PEA–H2O, respectively, are displayed, based on the calculated anharmonic vibrational frequencies and Raman intensities at the B3LYP level with a 6-311++G(d,p) basis set for the corresponding structures and convoluted with Lorentzian lines of full width half maximum of 0.5 cm−1. The calculated Raman spectra reflect the spectra corresponding to the electronic ground state, S0, structures of the most stable gauche conformers of the PEA monomer and of the PEA–H2O cluster, shown in panels (a) and (b), respectively. | ||
3.3 Determination of the structure corresponding to the measured ILSR spectrum of the PEA photofragment
By comparing the measured ILSR spectrum of the PEA fragment in Fig. 2(a), reflecting the PEA–H2O cluster (see below), to the calculated Raman spectra of the PEA(GI)-W, PEA(GIII)-W, PEA(GII)-W, PEA(AI)-W and PEA(AII)-W conformers, Fig. 2(b)–(f), respectively, it can be determined which of the calculated Raman spectra resembles mostly the measured one. It is clearly seen that by visually comparing the measured ILSR spectrum, Fig. 2(a), and the calculated Raman spectra, the best correspondence is obtained to the Raman spectrum related to the most stable gauche conformer PEA(GI)-W, shown in Fig. 2(b).Furthermore, in the second step, fitting quality indicators, based on the Euclidian distances were calculated. For the conformers PEA(GI)-W, PEA(GIII)-W, PEA(GII)-W, PEA(AI)-W and PEA(AII)-W the indicators were found to be 8.07, 11.9, 10.7, 11.6 and 13.4, respectively. Since a lower distance, means greater similarity between the measured and a specific calculated spectrum, it is seen that the spectrum corresponding to the PEA(GI)-W structure best fits the measured spectrum, significantly better than the spectra of the other conformers. This implies that the measured ILSR spectrum of the PEA fragment corresponds to the most stable conformer, where the amino hydrogen of the side chain weakly interacts with the aromatic π cloud. This conformation is related to the most highly populated conformer of the PEA monomer with the H2O HB directed to the nitrogen lone pair.
3.4 The PEA monomer and the PEA photofragment
Fig. 3(a) and (b) include, at the bottom of the panels, the calculated Raman spectra, corresponding to the electronic ground state, S0, structures of the most stable gauche conformers of PEA,18 and of PEA–H2O, namely PEA(GI)-W, following the above mentioned structure identification. In fact, the measured ILSR spectra indicate that the application of the SRS beams, i.e., ωp and tunable ωS, prior to the UV beam, which is parked on transitions corresponding to the PEA conformer, or to the PEA and CH2NH2–H2O photofragments in the R2PI spectra allows excitation of vibrational transitions, whenever the frequency differences, ωp − ωS, match vibrational frequencies with high enough Raman activities. These SRS transitions reduce the population of PEA molecules, or of PEA–H2O clusters in the vibrational ground state, decreasing the number of ions and thus allowing generation of the measured ILSR spectra.Comparison of the measured photofragment ILSR spectra of PEA [Fig. 3(b)] and of CH2NH2–H2O [Fig. 3(c)] shows that they resemble very much, although the signal to noise ratio in the latter is poorer. On the other hand, some differences between the ILSR spectra of the PEA monomer [Fig. 3(a)] and of the photofragments [Fig. 3(b) and (c)] are observed. These are related to some modes that appear only in a specific spectrum, belonging to one of the species, while others that seem to be only slightly perturbed and thus only somewhat shifted in frequencies (see Table S1 of the ESI).†
The most pronounced difference is the appearance of two new clear features in the high frequency range of the ILSR spectrum of the PEA photofragment, at 3717 and 3329 cm−1, characterized by full width half maxima (FWHM) of 2.2 and 10 cm−1, respectively. These two features are related to the fundamental O–H stretches of the unbound and bound water OH groups, respectively, and are close to the values observed by fluorescence-dip infrared spectroscopy in the tryptamine–H2O cluster, 3716 and 3340 cm−1.29
Comparison of the structures in Fig. 3(a) and (b), shows that the addition of water does not perturb the original conformation of the PEA monomer, but rather the PEA is approached by the water molecule with the hydrogen atom aiming toward the lone electron pair of the nitrogen atom. It is interesting to note that the structure of PEA–H2O is completely different from that obtained very recently for the hydrated cluster of protonated PEA, i.e., H+PEA–H2O, due to the large variation in charge distributions.30 In this case, the infrared photodissociation (IRPD) spectra suggested two classes of isomers, whereas in the first one the H2O was inserted into the NH+–π bond of bare H+PEA and was stabilized by two intermolecular NH+–O and OH–π interactions, while in the slightly less stable second class of isomers the H2O ligand acted as a an acceptor in a NH+–O HB to one of the free NH bonds of the NH3+ group.
In the formed PEA–H2O complex, the OH group of water can be considered as a HB donor31 to the lone pair of nitrogen. The formation of the HB causes linewidth broadening of this feature and a large red shift (∼325 cm−1) of the stretching mode of the bound OH, relative to that of the H2O monomer.32 The large shift toward lower wavenumber, indicates a strong interaction upon HB formation, leading to weakening of the O–H covalent bond and therefore to a force constant of O–H⋯N considerable smaller than that of the non-bonded O–H. The strength of this HB is also supported by the calculated binding energy, obtained at the MP2/6-311++G(d,p) level of theory [with zero-point vibrational energy corrections from B3LYP/6-311++G(d,p)] of 2728 cm−1 and that estimated by the basis set superposition error (BSSE) correction via counterpoise correction,33,34 which was found to be 2885 cm−1.
Moreover, comparison of the calculation results, shown in Fig. 3(a) and (b), confirms the appearance of these two bands upon complexation of PEA with water, showing fairly good agreement with the observed spectra and indicating that the high frequency peak corresponds to the non-bonded fundamental O–H stretch, while the low one is that of the bound O–H stretch. Even more so, the large red shift of the bound O–H stretch pushed it beyond the N–H stretching region, being definitely consistent with the calculated Raman spectrum of the cluster.
As for the N–H stretching modes of PEA at 3414 and 3346 cm−1 and the C–H stretching modes of the ring and of the ethylamino group, above and below 3000 cm−1, respectively, it can be clearly seen that they undergo only a little or no shift in wavenumber when the cluster is formed, agreeing quite well with the calculated values. Similar behavior is also observed in the lower frequency range that includes bending modes of the PEA and of the water as well as cooperative modes of the former. This is since the vibration modes of PEA–H2O appear close to the corresponding PEA modes, or they are shifted to positions of other bands.
Nevertheless, when an expanded portion of the measured ILSRS spectra of PEA and of the PEA photofragment is shown in Fig. 4(a) and (b), respectively, it can be clearly seen that some differences occur. For example in the region below 1210 and around 880 cm−1 different patterns are observed in the spectra of the two species. Furthermore, the band at ∼761 cm−1 is clearly seen in the measured ILSR spectrum of PEA, but missing in that of the PEA photofragment ILSR spectrum. This behavior is also observed in the calculated spectra and it turns out that this vibrational mode, corresponding to NH2 wagging, is strongly hindered by the presence of the water molecule in the cluster.
 | ||
| Fig. 4 An expanded portion of the ionization-loss stimulated Raman spectra obtained when the UV laser is parked on the C (a) and E (b) features related to 2-phenylethylamine (PEA) in Fig. 1(a). Also shown at the bottom of panels (a) and (b) are the theoretical spectra of PEA and of PEA–H2O, respectively, based on the calculated anharmonic vibrational frequencies and Raman intensities at the B3LYP level with a 6-311++G(d,p) basis set for the most stable conformers and convoluted with Lorentzian lines of full width half maximum of 0.5 cm−1. The black trace in panel (b) shows a zoomed portion of the spectrum. | ||
It is important to note that although photofragment ILSR spectroscopy was used, the measured spectra of the PEA and CH2NH2–H2O fragments are actually reminiscent of the signature of PEA–H2O cluster. This could be understood by considering that the SRS vibrational excitation, which depleted some of the population of the ground vibrational states occurred prior to the UV excitation that fragmented the parent ion, thus allowing monitoring of the characteristic spectrum of the PEA–H2O cluster.
4 Summary
The use of photofragment ILSRS together with DFT calculations allowed studying the most stable PEA–H2O cluster in the gas phase. As shown, this method provides the opportunity to measure the characteristic ILSR spectral signatures of clusters that their parent ions could not be directly observed by R2PI, due to the fragmentation occurring in the ionic state. It is anticipated that this technique can be used for measurement of the spectra of photofragments resulting from other molecules or clusters and to allow their structural assignment.Comparison of the spectra of PEA and of PEA–H2O photofragments with the corresponding calculated spectra and particularly the analysis of the features representing the O–H stretch and other modes in the low frequency range showed that the lowest energy conformation of PEA in its water cluster is similar to bare PEA. This structure is characterized by an ethylamino tail, folded in a gauche arrangement, with the amino hydrogen weakly interacting with the aromatic π cloud and with the hydrogen of H2O, pointing toward the nitrogen lone pair and forming a strong HB. The addition of the water molecule does not affect too much the PEA structure and even the flexible tail remains basically unaltered.
Acknowledgements
We thank Otto Dopfer (Technical University of Berlin) for fruitful discussions. Financial support of this research by the German-Israeli Foundation for Scientific Research and Development (G.I.F) under Grant no. 1164-158.5/2011 and by the Israel Science Foundation (ISF) founded by The Israel Academy of Science and Humanities is gratefully acknowledged.References
- J. P. Simons, C. R. Chim., 2003, 6, 17–31 CrossRef CAS.
- J. P. Schermann, Spectroscopy and Modeling of Biomolecules, Elsevier, Amsterdam, The Netherlands, 2008 Search PubMed.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1–18 RSC.
- J. P. Simons, Mol. Phys., 2009, 107, 2435–2458 CrossRef CAS.
- T. Ebata, A. Fujii and N. Mikami, Int. Rev. Phys. Chem., 1998, 17, 331 CrossRef CAS.
- T. S. Zwier, J. Phys. Chem. A, 2001, 105, 8827 CrossRef CAS.
- R. Weinkauf, J. P. Schermann, M. S. de Vries and K. Kleinermanns, Eur. Phys. J. D, 2002, 20, 309–316 CrossRef CAS.
- M. S. de Vries and P. Hobza, Annu. Rev. Phys. Chem., 2007, 58, 585–612 CrossRef CAS PubMed.
- M. Mons, I. Dimicoli and F. Piuzzi, Int. Rev. Phys. Chem., 2002, 21, 101–135 CrossRef CAS.
- S. J. Martinez III, J. C. Alfano and D. H. Levy, J. Mol. Spectrosc., 1993, 158, 82–92 CrossRef.
- S. Sun and E. R. Bernstein, J. Am. Chem. Soc., 1996, 118, 5086–5095 CrossRef CAS.
- J. Yao, H. S. Im, M. Foltin and E. R. Bernstein, J. Phys. Chem. A, 2000, 104, 6197–6211 CrossRef CAS.
- J. A. Dickinson, M. R. Hockridge, R. T. Kroemer, E. G. Robertson, J. P. Simons, J. McCombie and M. Walter, J. Am. Chem. Soc., 1998, 120, 2622–2632 CrossRef CAS.
- M. R. Hockridge and E. G. Robertson, J. Phys. Chem. A, 1999, 103, 3618–3628 CrossRef CAS.
- R. Weinkauf, F. Lehrer, E. W. Schlag and A. Metsala, Faraday Discuss., 2000, 115, 363–381 RSC.
- P. D. Godfrey, L. D. Hatherley and R. D. Brown, J. Am. Chem. Soc., 1995, 117, 8204–8210 CrossRef CAS.
- J. C. López, V. Cortijo, S. Blanco and J. L. Alonso, Phys. Chem. Chem. Phys., 2007, 9, 4521–4527 RSC.
- A. Golan, N. Mayorkas, S. Rosenwaks and I. Bar, J. Chem. Phys., 2009, 131, 024305 CrossRef PubMed.
- S. Melandri, A. Maris, B. M. Giuliano, L. B. Favero and W. Caminati, Phys. Chem. Chem. Phys., 2010, 12, 10210–10214 RSC.
- G. V. Hartland, B. F. Henson, V. A. Venturo, R. Hertz and P. M. Felker, J. Opt. Soc. Am. B, 1990, 7, 1950–1959 CrossRef CAS.
- N. Mayorkas, I. Malka and I. Bar, Phys. Chem. Chem. Phys., 2011, 13, 6808–6815 RSC.
- N. Mayorkas, S. Izbitski, A. Bernat and I. Bar, J. Phys. Chem. Lett., 2012, 3, 603–607 CrossRef CAS; N. Mayorkas, A. Bernat, S. Izbitski and I. Bar, J. Chem. Phys., 2013, 138, 124312 CrossRef PubMed.
- M. Epshtein, A. Portnov, R. Kupfer, S. Rosenwaks and I. Bar, J. Chem. Phys., 2013, 139, 184201 CrossRef PubMed.
- Avogadro: an open-source molecular builder and visualization tool, Version 1.00, see http://avogadro.openmolecules.net/ CAS; M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. J. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- J. R. Clarkson, J. M. Herbert and T. S. Zwier, J. Chem. Phys., 2007, 126, 134306 CrossRef PubMed.
- A. Bouchet, M. Schütz, and O. Dopfer, unpublished.
- S. Melandri, Phys. Chem. Chem. Phys., 2011, 13, 13901–13911 RSC.
- T. Shimanouchi, Tables of Molecular Vibrational Frequencies Consolidated Volume I, National Bureau of Standards, 1972, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P.J. Linstrom and W.G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD20899, 3 November 2014, http://webbook.nist.gov Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10493e |
| This journal is © The Royal Society of Chemistry 2014 |