
2-Hydroxyarylimidazole-based colorimetric and ratiometric fluoride ion sensors†
Abstract
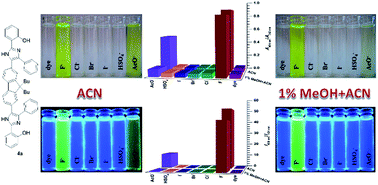
Two novel 2-hydroxyarylimidazoles were developed as colorimetric and ratiometric sensors for the naked-eye detection of fluoride anions. The discriminating absorption and fluorescence responses to fluoride in acetonitrile as well as a methanol–acetonitrile solvent system were observed. The bathochromic optical changes are attributed to the deprotonated species evidenced from 1H NMR titrations. Their selectivity for fluoride over acetate was successfully established in the methanol–acetonitrile mixture. A fluorene-bridged imidazole dimer, 4a demonstrated better response for fluoride than acetate in the neat acetonitrile. Moreover, the sensor 4a exhibited a larger ratio of fluorescence intensity response (I489/I431 = 51) than 4b (I521/I448 = 18) for fluoride in acetonitrile.
 Please wait while we load your content...
Please wait while we load your content...

