
Ligand substitution and electron transfer reactions of trans-(diaqua)(salen)manganese(iii) with oxalate: an experimental and computational study†
Abstract
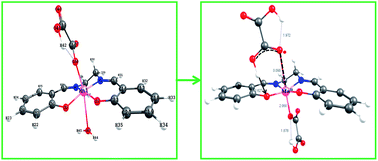
The trans-MnIII(salen)(OH2)2+ undergoes reversible aqua ligand substitution by HOX− (H2salen = N,N′-bis(salicylidene)ethane-1,2-diamine; HOX− = −O–COCO2H) with k1/dm3 mol−1 s−1 (k−1/s−1) = 11.8 ± 0.7 (0.255 ± 0.02), ΔH≠/kJ mol−1 = 54.6 ± 0.8 (64.2 ± 6.7), ΔS≠/J K−1 mol−1 = −41.2 ± 2.6 (−40.8 ± 22.7) at 25.0 °C and I = 0.3 mol dm−3. The low values of the activation enthalpy and nearly the same and negative values of the activation entropy are ascribed to an associative transition state for this interchange process (Ia mechanism). The redox reaction that follows involves several paths and the products are MnII and CO2 identified by ESR spectroscopy and conventional test, respectively. The rate retardation by acrylamide monomer with no perceptible polymerization during the course of the redox reaction supports the involvement of the radical intermediate, C2O4−˙ (= CO2 + CO2−˙) which succeeds in reducing MnIII species much faster than the dimerisation of its congener, CO2−˙ in keeping with the stoichiometry, |[ΔMnIII]/Δ[OX]| = 2. The trans-[MnIII(salen)(OH2)(HOX) and its conjugate base, trans-MnIII(salen)(OH2)(OX)− are virtually inert to intramolecular reduction of the MnIII centre by the bound oxalate species but undergo facile electron transfer by H2OX, HOX− and very slowly by OX2− following the reactivity sequence, kH2OX > kHOX ⋙ kOX and featuring second order kinetics. The rate retardation by the anionic micelles of SDS (sodium dodecyl sulfate) and rate enhancement by N3− provide supportive evidence in favor of the proposed mechanistic pathways. The structure optimization of trans-MnIII(salen)(OH2)(HOX) (A), trans-MnIII(salen)(HOX)2− (B), trans-MnIII(salen)(OH2)(OX)− (C), trans-MnIII(salen)(OH2)(H2OX)+ (E1), and trans-MnIII(salen)(HOX)(H2OX) (E2) {all high spin MnIII(d4)} by Density Functional Theory (DFT) reveals that the structural trans-effect of the unidentately bonded OX2− in C is the strongest and MnIII assumes five coordination with the H2O molecule (displaced from the MnIII centre), hydrogen bonded to the phenoxide oxygen moiety. The computational study highlights different modes of H-bonding in structures A–E. The activation parameters for the redox reactions, A + HOX− and A + H2OX, ΔH≠/kJ mol−1 (ΔS≠/J K−1 mol−1): 42.5 ± 6.2, (−106 ± 20) and 71.7 ± 7.7 (+12 ± 25), respectively, are indicative of different degrees of ordering and reorganization of bonds as expected in the case of a proton coupled electron transfer (PCET) process.
 Please wait while we load your content...
Please wait while we load your content...

