
Graphene oxide incorporated collagen–fibrin biofilm as a wound dressing material
Abstract
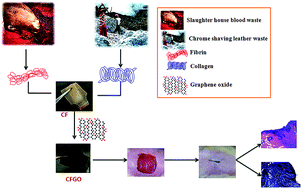
Functionalized graphene oxide offers many advantages as a biomaterial in various biomedical applications. In the present study, graphene oxide (GO) was incorporated in the collagen–fibrin composite film (CFGO) and used as wound dressing material in both in vitro and in vivo studies. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using NIH 3T3 cells revealed that CFGO film is biocompatible. CFGO was also used as a wound dressing material on the experimental wounds of rats. Histopathological, biochemical and hematology studies have shown that the CFGO treated wounds healed faster than control and CF treated wounds. Based on the results, CFGO can be used as a wound dressing material in small and large animals.
 Please wait while we load your content...
Please wait while we load your content...

