
Strength and failure behavior of a graphene sheet containing bi-grain-boundaries†
Baocheng Yang‡
a,
Shuaiwei Wang‡a,
Yanzhen Guoa,
Jinyun Yuana,
Yubing Sia,
Shouren Zhanga and
Houyang Chen*b
aInstitute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou, Henan 450006, China
bDepartment of Chemical and Biological Engineering, State University of New York at Buffalo, Buffalo, New York 14260-4200, USA. E-mail: hchen23@buffalo.edu
First published on 16th October 2014
Abstract
By using molecular dynamics simulations, the mechanical properties and failure mechanisms of a graphene sheet containing bi-grain-boundaries were examined. The results reveal that both temperature and density of defects play central roles in the mechanical characteristics of graphene containing bi-grain-boundaries. By increasing the temperature, the tensile strength and fracture strain significantly decrease. The graphene containing high density defects is much stronger than that containing lower density defects. The dependence of Young's modulus on temperature is also investigated. The results also show that the failure processes of graphene sheets containing bi-grain-boundaries are dominated by brittle cracking.
I. Introduction
Graphene has attracted great attention due to its outstanding electronic,1 thermal,2 and mechanic properties,3 etc. Chemical vapor deposition (CVD) is one of the popular methods for fabricating graphene films.4–7 Generally, the graphene sheets fabricated by CVD contain impurities and/or grain boundaries (GBs). These GBs alter the material characteristics. For example, the electron mobility in graphene was depended on the boundaries.8Understanding the effect of these defects on mechanical properties is important for their applications in the graphene-based devices. By employing scanning tunneling microscopy (STM), Simonis et al.9 identified that the GBs in the graphite contain pentagon–heptagon pairs. Periodic arrays of line defect rings composed by pentagon–heptagon10 or pentagon–octagon11 pairs have been observed by transmission electron microscopy (TEM). By employing atomic force microscopy (AFM), Huang et al.12 measured the strength of graphene sheets, and discovered that the grain boundaries can change the mechanical strength drastically.
Theoretically, the electronic, transport, mechanical, and magnetic properties of graphene sheet with one grain boundaries (one-GBs) are examined by using the first principle calculations with the density functional theory (DFT) and/or (semi) empirical simulation. Yazyev and Louie13 determined that the dependence of the electronic structures of the graphene sheets on the dislocations and one-GBs. Additionally, two distinct transport behaviors (high transparency and perfect reflection of charge carriers over large energy ranges) in the large-area graphene were found.14 Grantab et al.15 reported that graphene with the large-angle tilt boundaries were much stronger than those with a smaller angle tilt boundaries, and the fracture bonds were dependent on both the orientation of the graphene and the loading direction. Zhang et al.16 found that the Stone–Wales transformation was the major failure mechanism of one-GBs, and the fracture site began either on the boundary line or inside the domain. Meanwhile, the same group investigated the electronic transport of the graphene sheet, and found that the mismatch angle of GB determined electronic transport.17 In the past, most of reports were studied concerning the graphene with one grain boundary. However, the grain boundaries generated in experiments are complex.4,8,18–20 To mimic more complicated GBs in graphene, in this paper, we focus on graphene with two grain boundaries (denoted as bi-grain-boundary), in which the atom structures are more complicated than that in graphene with one-grain-boundary. Regarding the graphene containing bi-grain-boundary (bi-GB), the properties are rare. Márk et al.21 used the wave packet dynamical simulation technique to investigate the behavior of GBs which contains a pair of parallel GBs, and they found that the graphene ribbon confined between the GBs may behave like a channel for charge carries, and this would be created in the nanoscale electronic waveguides. Lee et al.22 investigated the motion and annihilation of bi-GB in graphene using tight-binding molecular dynamics simulation and ab initio local density approximation. They found that those properties of graphene are dependent on the nature of the grain boundaries.
Employing the molecular dynamics (MD) simulations in this work, we examine the mechanical properties and failure processes of graphene containing bi-grain-boundary. Dependence of tensile strength and Young's modulus on the temperature and density of defects have been investigated. In addition, the failure mechanism was discussed. This is the first instance in which a report concerning the mechanical properties of graphene containing bi-GB was accomplished.
II. Model and methodology
In the present paper, three mismatched zigzag-oriented (denoted by ZZ1, ZZ2, and ZZ3) and three mismatched armchair-oriented (AC4, AC5, and AC6) graphene sheets were examined (see Fig. 1). Each graphene sheet possesses two grain boundaries (bi-grain-boundary), which stitched together with pentagon and heptagon (5–7 rings) in the opposite direction, have two mismatch angles. The distance between the two grain boundaries is approximately 9.0 Å. The misorientation angles are (⊤5.5°, ⊥5.5°), (⊤13.2°, ⊥13.2°), (⊤21.8°, ⊥21.8°), (⊤17.9°, ⊥17.9°), (⊤21.8°, ⊥21.8°), and (⊤27.8°, ⊥27.8°) for ZZ1, ZZ2, ZZ3, AC4, AC5 and AC6, respectively. The size of graphene was 25.0 × 25.0 (nm)2. In addition, two 5.0 Å wide strips at each edge of x-axis were constrained against motion. There are free edges and periodic boundary condition in the x- and y-direction, respectively. Simulations were performed by large-scale atomic/molecular massively parallel simulator (LAMMPS).23 The adaptive intermolecular reactive empirical bond-order (AIREBO) potential,24 which has been proven to accurately reproduce the mechanical properties of graphene containing grain boundaries,15,16,25 is used to account for the carbon–carbon interactions. The minimum cutoff distance 1.92 Å is employed in the AIREBO potential. | ||
| Fig. 1 The zigzag-oriented (a–c) and armchair-oriented (d–f) graphene contain bi-grain-boundary with various mismatch angles. The pentagons and heptagons are highlighted in light gray. (a) ZZ1: (⊤5.5°, ⊥5.5°); (b) ZZ2: (⊤13.2°, ⊥13.2°); (c) ZZ3: (⊤21.8°, ⊥21.8°); (d) AC4: (⊤17.9°, ⊥17.9°); (e) AC5: (⊤21.8°, ⊥21.8°); (f) AC6: (⊤27.8°, ⊥27.8°). | ||
In order to obtain optimized structures, the graphene containing bi-grain-boundary was relaxed by using the conjugate gradient method. Then the simulation with NVT ensemble is employed in the Nosé–Hoover thermal bath,26 coupling for 2.5 ns with a time step of 1 fs at 300 K. After the equilibrium states are achieved, tensile tests are loaded in the x-direction (normal to the direction of the grain boundary) until complete failure is achieved. The engineered strain rate is 0.0004 ps−1 and the increment are applied every 0.1 ps.
To understand mechanical properties in detail, the stress–strain curves, the Young's modulus E, the fracture strain, and the fracture strength were examined as well. In order to calculate the stress–strain curves during loading, the per-atom stress on each carbon atom is calculated according to the following virial stress equation27–29
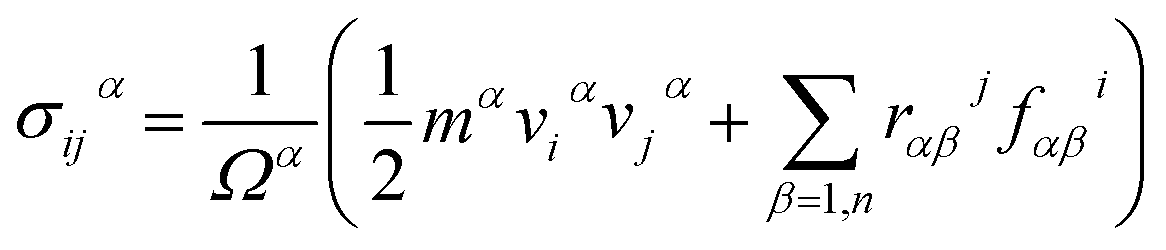 | (1) |
III. Results and discussion
First, we would like to verify the simulation method and the force field employed. The stress–strain curves of pristine graphene and graphene containing one-grain-boundary under uniaxial tension in both zigzag and armchair directions are shown in Fig. 2. The trend of the stress–strain curves are the same as the previous DFT and MD results.15,25 In addition, for the pristine graphene, one can obtain the tensile strength of 113.11 GPa in the zigzag direction (see Table 1) and 95.13 GPa in the armchair direction (see Table 1), respectively, which are in reasonable agreement with the values of 118.05 GPa (zigzag oriented graphene) and 97.80 GPa (armchair oriented graphene) from previous DFT calculations.25 For graphene containing one grain boundary, the tensile strength and fracture strain (see Table 1) are 78 GPa and 0.1243 for angle of 5.5°, respectively, agreeing with the results (79 GPa, 0.1125)25 and (71 GPa, 0.1047)15 of the same angle of the one-GB from previous MD studies. Our results suggest that Young's modulus of pristine graphene are 919.88 GPa in the armchair direction (see Table 1) and 761.29 GPa in the zigzag direction (see Table 1), which are consistent with results from both the experiment (1 TPa)3 and MD simulations (0.95 TPa)31 in the armchair direction. Moreover, the Young's modulus are smaller variations of the one GBs. The present results are in excellent agreement with the results from both the MD simulations and experiments, indicating that the methodology and interatomic potential that are adopted can provide a reliable description of the mechanical properties of graphene containing grain boundaries. | ||
| Fig. 2 The stress (σ)–strain (ε) curves of pristine graphene and graphene containing one-grain-boundary with various tilt angles under tensile loading in the armchair (a) and zigzag (b) direction. | ||
| Pristine graphene | Graphene containing one-grain-boundary | |||||||
|---|---|---|---|---|---|---|---|---|
| Zigzag | Armchair | Zigzag | Armchair | |||||
| 5.5° | 13.2° | 21.8° | 17.9° | 21.8° | 27.8° | |||
| a Ref. 25.b Ref. 15. | ||||||||
| σc (GPa) | 113.11 | 95.13 | 78.22 | 82.92 | 87.50 | 60.43 | 70.94 | 80.74 |
| 118.05a | 97.80a | 79.08a | 86.39a | 100.63a | 57.72a | 68.2a | 97.44a | |
| 71.39b | 75.11b | 94.10b | 49.02b | 57.05b | 93.92b | |||
| εf | 0.2774 | 0.1774 | 0.1243 | 0.1325 | 0.1450 | 0.0848 | 0.1040 | 0.1237 |
| 0.2785a | 0.1792a | 0.1125a | 0.1275a | 0.1796a | 0.0687a | 0.0853a | 0.1683a | |
| 0.1047b | 0.1102b | 0.1497b | 0.0664b | 0.0774b | 0.1493b | |||
| E (GPa) | 761.29 | 919.88 | 771.84 | 777.47 | 810.50 | 827.86 | 831.81 | 833.17 |
The dependence of mechanical properties on bi-GB in graphene were further examined. The stress–strain curves, the corresponding tensile strength, and the fracture strain for bi-GB graphene in the armchair and zigzag direction with various tilt angles are shown in the Fig. 3a–c, respectively. As the tilt angle increases, the tensile strength and the fracture stain increases significantly in both armchair and zigzag graphene with bi-GBs. In addition, one can conclude that the tensile strength of graphene containing bi-GB is lower than that of pristine graphene. For example, the tensile strength 88.85 GPa for ZZ3 (⊤21.8°, ⊥21.8°), which is lower than that in the pristine graphene in zigzag (95.13 GPa) direction, is somewhat higher than that (87.50 GPa) in the same angle of one-GB (see Table 1). The fracture strain for AC4 (⊤17.9°, ⊥17.9°), AC5 (⊤21.8°, ⊥21.8°) and AC6 (⊤27.8°, ⊥27.8°) are 0.0836, 0.0978 and 0.1228, respectively, which is closer to the values in the same angle of one-GBs. Generally, the graphene containing bi-GBs exhibit similar mechanical properties to the graphene containing one-GB.
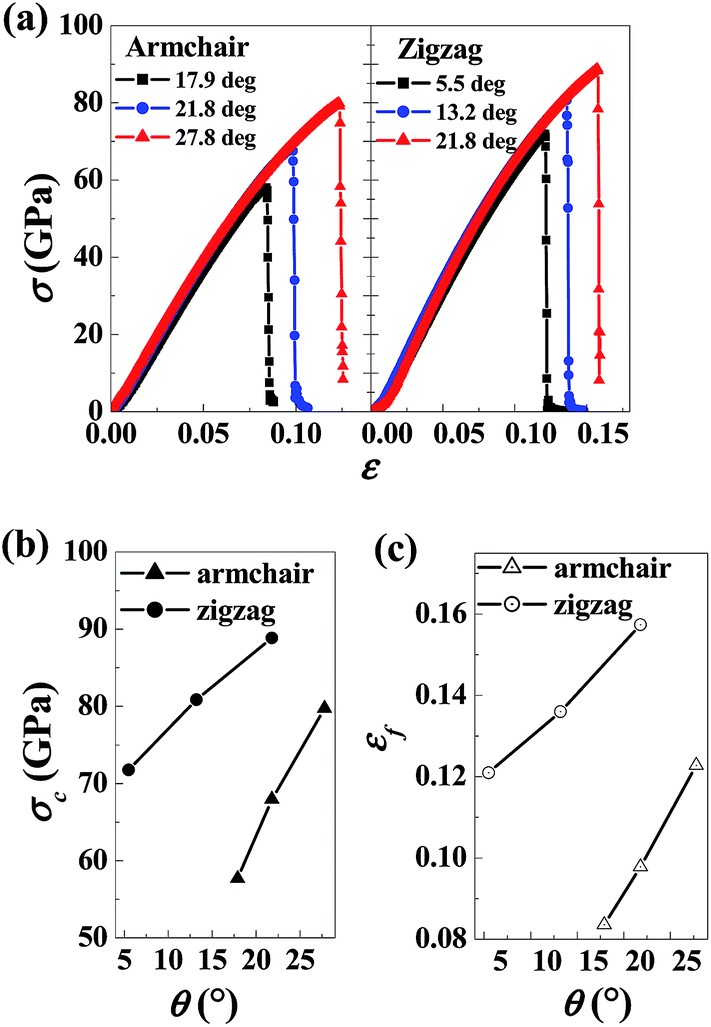 | ||
| Fig. 3 (a) Stress (σ) versus strain (ε), (2) fracture strength (σc) versus tilt angles (θ) and (c) fracture strain (εf) versus tilt angles (θ) of graphene containing bi-grain-boundary in both armchair and zigzag directions. | ||
To examine the effects of temperature on the mechanical properties of graphene containing bi-GBs, the dependence of the tensile strength and the fracture strain on temperature from 1 K to 1200 K are plotted in Fig. 4. The tensile strength and fracture strain decrease significantly with the increase in temperature. For instance, the tensile strength 55 GPa of ZZ3 (⊤21.8°, ⊥21.8°) at 1200 K is approximately 41% and 38% lower than those at 1 K (94 GPa) and at room temperature (89 GPa), respectively. The fracture strain 0.089 at 1200 K is approximately 47% and 43% lower than those at 1 K (0.169) and at room temperature (0.158), respectively. As shown in Fig. 4a, the tensile strength 66 GPa of AC6 (⊤27.8°, ⊥27.8°) at T = 1200 K is approximate 26% and 21% lower than those at 1 K (89 GPa) and at room temperature (84 GPa), respectively. The fracture strain 0.116 of AC6 at 1200 K is approximate 30% and 22% lower than those at 1 K (0.166) and at room temperature (0.149). The dependence of the tensile strength and strain of the pristine graphene and of the graphene containing one-GBs on temperature has been found in MD simulation32,33 and DFT calculations.34 In addition, it is readily found that the graphene with higher density defects have a larger tensile strength and larger fracture strain than those with lower density defects. Comparing all the bi-GBs in the armchair direction, the tensile strength and fracture of AC6 (⊤27.8°, ⊥27.8°) is greater than those of AC4 (⊤17.9°, ⊥17.9°) and AC5 (⊤21.8°, ⊥21.8°). The tensile strength and fracture strain of bi-GBs both in the armchair and zigzag directions decrease at almost the same rate as the temperature increases. The increase in temperature causes strong vibrations between the atoms, which mainly cause the tendency of the decrease in tensile strength and fracture strain. The tendency of curves for graphene containing bi-GBs in our work is similar to those of one-GB.35,36
 | ||
| Fig. 4 The fracture strength (σc) and fracture strain (εf) of graphene sheet containing bi-grain-boundary as functions of temperature (T) with different tilt angles. (a) Square: AC4 (⊤17.9°, ⊥17.9°); triangle: AC5 (⊤21.8°, ⊥21.8°); circle: AC6 (⊤27.8°, ⊥27.8°); diamond: ZZ1 (⊤5.5°, ⊥5.5°), pentagram: ZZ2 (⊤13.2°, ⊥13.2°); hexagonal: ZZ3 (⊤21.8°, ⊥21.8°). The solid symbols and the open symbols are fracture strength (σc) and fracture strain (εf), respectively. | ||
The Young's modulus of graphene containing bi-GBs can be obtained from the stress–strain curve by using Hooke's law σ = Eε and they are presented in Fig. 5. Similar to the trend of fracture strain and tensile strength, for a selected bi-GB, the Young's modulus decreases with the increase in temperature. The Young's modulus decreases by 9.2%, 8.7% and 7.3% from 814 GPa, 872 GPa and 873 GPa at 1 K to 739 GPa, 796 GPa and 809 GPa at 1200 K for AC4 (⊤17.9°, ⊥17.9°), AC5 (⊤21.8°, ⊥21.8°) and AC6 (⊤27.8°, ⊥27.8°) in armchair direction (Fig. 5a), respectively. The Young's modulus of graphene containing bi-GBs in the armchair direction is somewhat lower than that in the pristine graphene.34 For the bi-GBs in the zigzag direction, the Young's modulus increases significantly with the increase of the tilt angle. Compared with the bi-GB in the zigzag direction, the Young's modulus with bi-GBs in the armchair direction is more sensitive to temperature. The softening tendency of Young's modulus vs. temperature is larger than those in pristine graphene.34 The reduced ratio of Young's modulus in this work is similar to that in the graphene containing one GB for the zigzag-oriented graphene from MD simulations.35
 | ||
| Fig. 5 The Young's modulus (E) versus temperature (T) with various tilt angles. (a) Square: AC4 (⊤17.9°, ⊥17.9°); triangle up: AC5 (⊤21.8°, ⊥21.8°); circle: AC6 (⊤27.8°, ⊥27.8°); pentagram: ZZ1 (⊤5.5°, ⊥5.5°), triangle down: ZZ2 (⊤13.2°, ⊥13.2°), diamond: ZZ3 (⊤21.8°, ⊥21.8°). | ||
In order to show the fracture process of the graphene containing bi-GBs, we analyze the first signs of failure during deformation perpendicular to the boundaries. As shown in Fig. 6, the fracture processes begin from the bonds (highlighted by a red circle) shared by the 7–6 rings. Similar behavior was found in the failure processes in graphene containing one-grain-boundary.15,36,37 It is obvious that the first break bonds are the identical ones for all six grain boundary angles. At low temperatures (e.g. T = 1 K), after the first broken bond occurs, a hole is quickly formed from the origin of the crack, and another bond breaks during other defects with further stretching (see Fig. S1†). At high temperatures (e.g. T = 1200 K), once the first broken bond occurs, several bonds break simultaneously (see Fig. S1†), then the cracks begin from these broken bonds.
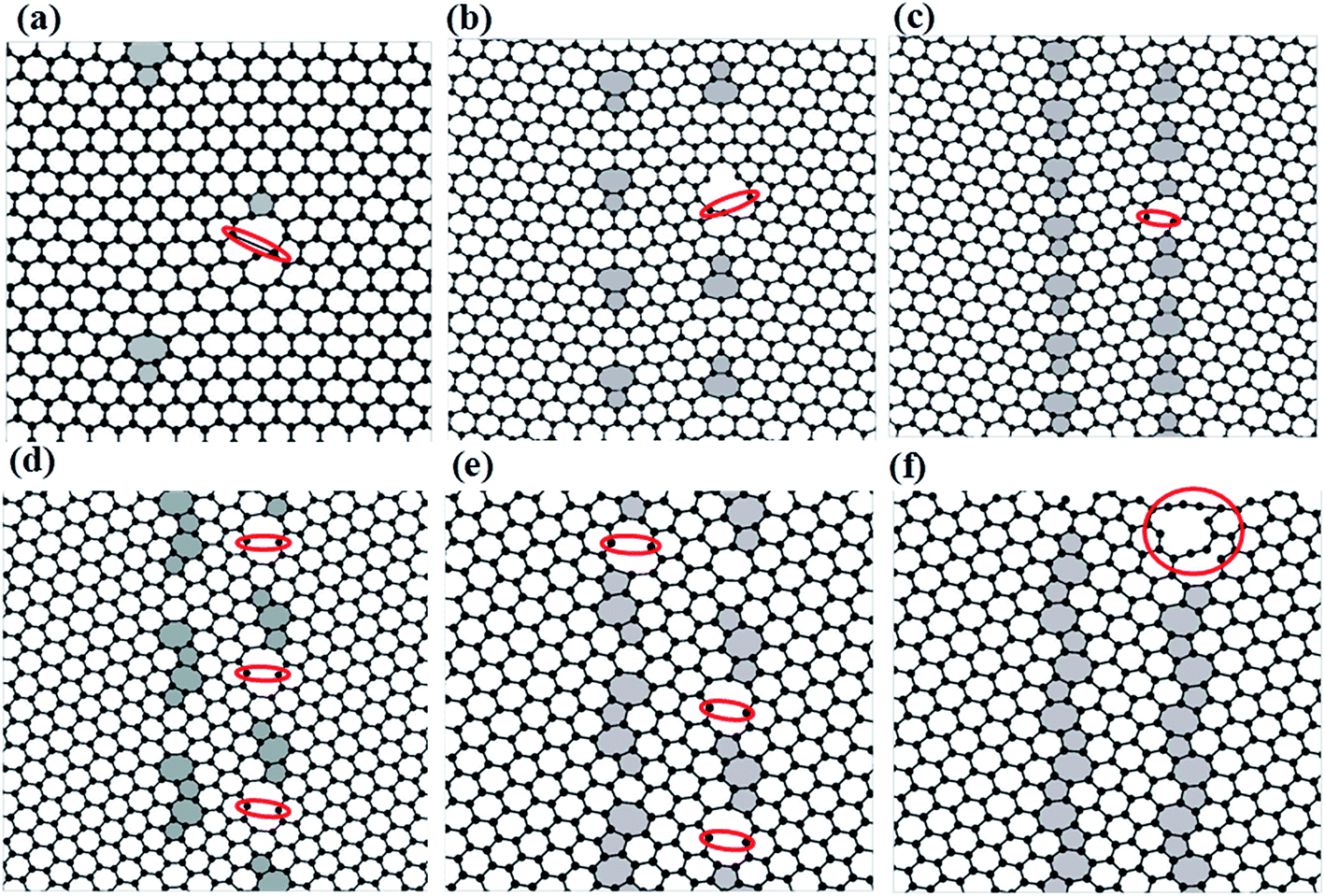 | ||
| Fig. 6 The initial stages of fracture sites in zigzag-oriented (a–c) and armchair-oriented (d–f) graphene sheets pulled perpendicular to the graphene containing bi-grain-boundary at T = 300 K. | ||
To further understand the mechanisms of the failure process for bi-GBs, the atomic stress distributions for AC4 (⊤17.9°, ⊥17.9°) are illustrated in Fig. 7. It had been found that the bonds shared by hexagon–heptagon rings (7–6 rings) in the grain boundary bear a much higher stress than the bond shared by hexagon–hexagon rings (Fig. 7a). With further stretching, the atoms on the hexagon–heptagon rings were found to exhibit a much higher stress than both the 6–5 ring in the grain boundary and the other atoms in the system (Fig. 7b). These phenomena are mainly due to the out-of-plane bucking array on the grain boundary after relaxation and the atoms in the out-of-plane bucking have little stress. Similar results have been found in one-GBs38 cases by using MD simulations and ab initio calculations. Furthermore, the critical stress is dependent on the loading direction. More fractures occur within the corresponding bonds which are shared by 7–6 ring (Fig. 7c). Before a complete fracture occurs, a short nanowire can be observed in the Fig. 7d. Thereafter, crack propagates along the grain boundary direction are generated. In short, the brittle cracking mechanism dominates the failure process in the bi-GBs. The same failure mechanism also can be found in graphene containing one-GBs16,33,39 and carbon nanotube.40–42 It should be mentioned that at high temperature, if the strain rate becomes small enough, the defects on the graphene may migrate and reconstruct which might introduce ductile behavior.
 | ||
| Fig. 7 Deformation processes and distribution stress field of AC4 (⊤17.9°, ⊥17.9°) at T = 300 K. (a) After relaxation (b) 3.6% strain (c) bond break at 6.8% strain (d) nanowire at 6.9% strain. The pentagons and heptagons are highlighted in light grey. | ||
IV. Conclusion
The mechanical properties and the failure processes of the graphene sheet containing bi-GBs at various temperatures (from 1 to 1200 K) are investigated by using MD simulations. The simulation results show that the tensile strength and fracture strain decrease significantly as temperature increases. By increasing the density of defect (i.e. increasing tilt angle of bi-GB), the tensile strength and fracture strain as well as the Young's modulus become larger. The brittle cracking mechanism dominates the failure process. The initial failure site is on the grain boundary line. The results may provide an insight to design graphene-based materials, and offer a failure mechanism of graphene containing bi-GB at the atomic scale.Acknowledgements
This work is supported by the Leading Talents for Zhengzhou Science and Technology Bureau (grant no. 131PLJRC649) and the program for University Innovative Talents of Science and Technology in Henan Province (grant no. 2012HASTIT036). We thank the High performance Computing Center of Huanghe Science and Technology College for the computer time provided.References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, science, 2008, 321, 385–388 CrossRef CAS PubMed.
- Q. Yu, L. A. Jauregui, W. Wu, R. Colby, J. Tian, Z. Su, H. Cao, Z. Liu, D. Pandey and D. Wei, Nat. Mater., 2011, 10, 443–449 CrossRef CAS PubMed.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2008, 9, 30–35 CrossRef PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung and E. Tutuc, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- L. Gao, J. R. Guest and N. P. Guisinger, Nano Lett., 2010, 10, 3512–3516 CrossRef CAS PubMed.
- P. Simonis, C. Goffaux, P. A. Thiry, L. P. Biro, P. Lambin and V. Meunier, Surf. Sci., 2002, 511, 319–322 CrossRef CAS.
- K. Kim, Z. Lee, W. Regan, C. Kisielowski, M. F. Crommie and A. Zettl, ACS Nano, 2011, 5, 2142–2146 CrossRef CAS PubMed.
- J. Lahiri, Y. Lin, P. Bozkurt, I. I. Oleynik and M. Batzill, Nat. Nanotechnol., 2010, 5, 326–329 CrossRef CAS PubMed.
- P. Y. Huang, C. S. Ruiz-Vargas, A. M. van der Zande, W. S. Whitney, M. P. Levendorf, J. W. Kevek, S. Garg, J. S. Alden, C. J. Hustedt, Y. Zhu, J. Park, P. L. McEuen and D. A. Muller, Nature, 2011, 469, 389–392 CrossRef CAS PubMed.
- O. V. Yazyev and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 195420 CrossRef.
- O. V. Yazyev and S. G. Louie, Nat. Mater., 2010, 9, 806–809 CrossRef CAS PubMed.
- R. Grantab, V. B. Shenoy and R. S. Ruoff, Science, 2010, 330, 946–948 CrossRef CAS PubMed.
- J. Zhang, J. Zhao and J. Lu, ACS Nano, 2012, 6, 2704–2711 CrossRef CAS PubMed.
- J. Zhang and J. Zhao, Carbon, 2013, 55, 151–159 CrossRef CAS PubMed.
- A. Capasso, E. Placidi, H. F. Zhan, E. Perfetto, J. M. Bell, Y. T. Gu and N. Motta, Carbon, 2014, 68, 330–336 CrossRef CAS PubMed.
- D. L. Duong, G. H. Han, S. M. Lee, F. Gunes, E. S. Kim, S. T. Kim, H. Kim, Q. H. Ta, K. P. So and S. J. Yoon, Nature, 2012, 490, 235–239 CrossRef CAS PubMed.
- P. Nemes-Incze, P. Vancsó, Z. Osváth, G. I. Márk, X. Jin, Y.-S. Kim, C. Hwang, P. Lambin, C. Chapelier and L. PéterBiró, Carbon, 2013, 64, 178–186 CrossRef CAS PubMed.
- G. I. Márk, P. Vancsó, P. Lambin, C. Hwang and L. P. Biró, J. Nanophotonics, 2012, 6, 061718 CrossRef PubMed.
- G.-D. Lee, E. Yoon, C.-Z. Wang and K.-M. Ho, J. Phys.: Condens. Matter, 2013, 25, 155301 CrossRef PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- S. J. Stuart, A. B. Tutein and J. A. Harrison, J. Chem. Phys., 2000, 112, 6472–6486 CrossRef CAS PubMed.
- Y. Wei, J. Wu, H. Yin, X. Shi, R. Yang and M. Dresselhaus, Nat. Mater., 2012, 11, 759–763 CrossRef CAS PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef.
- Z. Basinski, M. Duesbery and R. Taylor, Can. J. Phys., 1971, 49, 2160–2180 CrossRef CAS PubMed.
- N. Chandra, S. Namilae and C. Shet, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 094101 CrossRef.
- S. Wang, B. Yang, S. Zhang, J. Yuan, Y. Si and H. Chen, ChemPhysChem, 2014, 15, 2749–2755 CrossRef CAS PubMed.
- R. Al-Jishi and G. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 4514–4522 CrossRef CAS.
- J.-W. Jiang, J.-S. Wang and B. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 113405 CrossRef.
- H. Zhao and N. Aluru, J. Appl. Phys., 2010, 108, 064321 CrossRef PubMed.
- H. Zhang, Z. Duan, X. Zhang, C. Liu, J. Zhang and J. Zhao, Phys. Chem. Chem. Phys., 2013, 15, 11794–11799 RSC.
- T. Shao, B. Wen, R. Melnik, S. Yao, Y. Kawazoe and Y. Tian, J. Chem. Phys., 2012, 137, 194901 CrossRef PubMed.
- A. Cao and J. Qu, J. Appl. Phys., 2012, 112, 043519 CrossRef PubMed.
- L. Yi, Z. Yin, Y. Zhang and T. Chang, Carbon, 2013, 51, 373–380 CrossRef CAS PubMed.
- J. Han, S. Ryu, D. Sohn and S. Im, Carbon, 2014, 68, 250–257 CrossRef CAS PubMed.
- T.-H. Liu, G. Gajewski, C.-W. Pao and C.-C. Chang, Carbon, 2011, 49, 2306–2317 CrossRef CAS PubMed.
- Y. I. Jhon, Y. M. Jhon, G. Y. Yeom and M. S. Jhon, Carbon, 2014, 66, 619–628 CrossRef CAS PubMed.
- T. Dumitrică and B. I. Yakobson, Appl. Phys. Lett., 2004, 84, 2775–2777 CrossRef PubMed.
- T. Dumitrică, T. Belytschko and B. I. Yakobson, J. Chem. Phys., 2003, 118, 9485–9488 CrossRef PubMed.
- T. Dumitrica, M. Hua and B. I. Yakobson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6105–6109 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra10126j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |