
Ligand-stabilized CdSe nanoplatelet hybrid structures with tailored geometric and electronic properties. New insights from theory†
A. Szemjonova,
T. Pauportéa,
S. Ithurriab,
N. Lequeuxb,
B. Dubertretb,
I. Ciofini*a and
F. Labat*a
aPSL Research University, Chimie-Paristech-CNRS, IRCP UMR, 8247, 11 Rue Pierre et Marie Curie, 75005 Paris, France
bLaboratoire de Physique et d'Etude des Matériaux, CNRS, ESPCI ParisTech, PSL Research University, UPMC Sorbonne University, 10 rue Vauquelin, 75231 Paris, France
First published on 17th October 2014
Abstract
Quasi-two dimensional CdSe nanoplatelets with a well-controlled thickness exhibit several advantageous properties for optical and opto-electronic application, such as in quantum dot sensitized solar cells. Due to the quantum confinement effects arising from their thickness of typically a few nanometers, the excitonic and charge carrier properties of these nanoobjects can be easily tuned by varying the number of monolayers they are composed of and the passivating ligands adsorbed on their surface. We have performed a density functional theory (DFT) investigation of the geometrical and electronic properties of non-stoichiometric CdSe zinc blende nanoplatelets with different thicknesses in the (100) direction, stabilized by various organic (HCOO−) and inorganic (SH− and OH−) ligands. The relaxation parameters and adsorption energies of the studied ligands on the polar zinc blende (100) surface have been calculated, along with the band gaps, band structures, density of states and a detailed Mulliken charge analysis of these hybrid nanostructures. The latter revealed a major electron transfer from the SH− ligand towards the surface of the nanocrystals, in line with what is observed from the orbital-projected density of states. CdSe zinc blende nanoplatelets of various thicknesses, stabilized by fatty acids, SH− and OH− ligands have also been synthesized, and their band gaps have been measured by absorption spectroscopy. A good agreement is found between the experimental and calculated values, especially for the evolution of the band gaps with the thickness of the nanoplatelets. Taken all together, the established theoretical model and computational approach can potentially serve as a powerful tool to provide a qualitative and quantitative description of the geometrical and electronic properties of quasi-two dimensional nonstoichiometric polar inorganic semiconductor materials, at low computational cost.
Introduction
Quantum dots (QDs) as light harvesters in quantum dot sensitized solar cells (QDSCs) have recently gained marked attention mainly because of the possibility of exceeding the Shockley–Queisser efficiency limit of p–n junction photovoltaic cells1 via multiple exciton generation (MEG)2 and of their tunable absorption spectrum.3 Due to their small size of typically a few nanometers,4 quantum confinement effects arise on their excitonic and charge carrier properties. As a result, their optical absorption bands exhibit a redshift as their dimensions are increased.4–6 Varying their size is thus one way to tune their absorption spectra. Another way of optimizing absorption edge (corresponding to the band gap) of QDs is to modify their surface electronic states, for example by adsorbing different ligands on their surface.7 In principle, band gap engineering makes thus possible to tune an optimal overlap between the absorption spectra of the light harvesting components of QDSCs and the solar spectrum.Cadmium selenide (CdSe) is one of the most extensively studied QD materials due to its easy and low-cost fabrication,8 photostability3 and possible multiple exciton generation.9 The wurtzite and zinc blende polymorphs are its two stable phases under ambient conditions.10 CdSe QDs of various shapes have been synthesized: spherical nanoparticles,11 nanorods,12 nanoribbons,13 tetrapods,14 nanosheets15 and nanoplatelets.5 As regards the latter, it is possible to fabricate CdSe zinc blende nanoplatelets (NPLs) with a well-defined thickness in the (100) direction, which is controlled at an atomic scale.16 These quasi two-dimensional nanocrystals exhibit several favorable properties for photovoltaic applications. They show much narrower excitonic absorption and photoemission bands5 and higher photoluminescence quantum yields17 than QDs and nanorods (NRs). Moreover, one can easily make use of the quantum confinement effects by varying their thickness.5 Finally, their flat shape and high surface area are also useful parameters for the design of photovoltaic devices. Taken all together, quasi two-dimensional (2D) nanocrystals exhibit promising properties for photovoltaics, and are interesting alternatives of QDs and NRs and other zero or one dimensional nanoobjects in QDSCs, such as nanorods and nanoparticles.
Typically, CdSe zinc blende nanoplatelets are capped with fatty acids.5,18,19 Organic capping agents of spherical CdSe quantum dots have been efficiently replaced by inorganic ligands (S2−, SH−, OH−, Te2−, TeH−) by ligand exchange reactions. These inorganic ligands have been shown to facilitate charge transfer compared to long-chain organic ones.11 The use of inorganic ligands can open up interesting possibilities for QDs of other shapes, such as the quasi-2D nanoplatelets in question as well.
Nonetheless, it is essential to have an accurate description of the electronic properties of QDs in order to understand charge transfer processes in QDSCs and the key factors governing their efficiency. However, the experimental measurement of two dimensional energetic properties like the surface formation energy is difficult and it can give ambiguous results because extremely clean materials are needed, in which the uniformity of the exposed surface is insured, which is often not the case.20 The difficulties related to experimentally obtaining an accurate description of the electronic and geometric properties at an atomistic scale further valorize the use of theoretical methods. Computational approaches have proven to be useful tools to simulate both bulk and surface properties of semiconductors,21,22 as well as adsorption of ligands or functional molecules on their surfaces.23–26 Among them, density functional theory (DFT) methods, especially when using hybrid functionals, were proven to provide an accurate description of both geometrical and electronic properties of semiconductor bulk crystals27,28 and surfaces28 at low computational cost.
In this work, a theoretical study of CdSe zinc blende nanoplatelets of different thicknesses in the (100) direction is presented. Three cases have been studied: (1) nanoplatelets stabilized by fatty acids, (2) thiols (SH−) (3) or hydroxide groups (OH−). A theoretical model of these ligand–nanocrystal hybrid systems has been built, and the geometrical and electronic properties of these nanocrystals were investigated in-depth within the DFT framework. The theoretical results obtained were validated by experimental measurements. More precisely, the band gaps of the synthesized nanoplatelets were measured by absorption spectroscopy.
Experimental section
Synthesis of CdSe nanoplatelets
The synthesis of 5, 7, 9 atomic layers thick nanoplatelets was based on ref. 5. In this work, one monolayer (ML) of a nanoplatelet stands for an atomic layer composed exclusively of cadmium or selenium atoms in the same 〈100〉 plane, as depicted in Fig. 1. As an example, in case of 9 layers thick nanoplatelets, 0.3 mmol of Cd(myristate)2, 0.15 mmol of selenium mesh and 10 ml of octadecene were degassed for 20 minutes in a three neck flask. Then, under argon flow, the temperature was set at 240 °C. When the mixture turned orange (around 200 °C), 0.2 mmol of Cd(Ac)2·2H2O were introduced through a neck. After 5 minutes at 240 °C, the mixture was quenched, and 2 ml of oleic acid and 35 ml of hexane were added. The nanoplatelets were separated by centrifugation and were resuspended in 10 ml of hexane. | ||
| Fig. 1 TEM views of (a) 9 and (b) 13 atomic layers thick CdSe nanoplatelets. The scale bar length is 20 nm. (c) Side views of the alternate stacking of positively (+ sign) and negatively (− sign) charged planes in a CdSe zinc blende five layers thick slab exposing its Cd-terminated (100) surface. Cd atoms are presented as grey spheres, Se atoms as yellow spheres. Blue arrows show the dipole momenta arising from the dangling bonds of the surface Cd atoms, the vector sum of which is represented by a red arrow. | ||
The synthesis of 11 atomic layers thick nanoplatelets was based on ref. 17. In a three neck flask, 0.3 mmol of Cd(myristate)2 and 10 ml of octadecene were degassed for 20 minutes. Then, under argon flow, the temperature was set at 240 °C and a suspension of 0.15 mmol of selenium mesh sonicated in 1 ml of octadecene was injected. After 1 minute, 0.4 mmol of Cd(Ac)2·2H2O were introduced through a neck. After 10 minutes at 240 °C, the mixture was quenched and 2 ml of oleic acid, and 35 ml of hexane were added. The nanoplatelets were separated by centrifugation and were resuspended in 10 ml of hexane.
The obtention of 13 atomic layers thick nanoplatelets was inspired by ref. 29. The selenium precursor was produced in the following way. In a glove box, 0.15 mmol of selenium mesh were introduced in 1 ml of N-methylformamide, then 0.3 mmol of NaBH4 dissolved in 0.5 ml of N-methylformamide were dropwise added in order to reduce Se to Se2−, The flask was stirred open (in order to release H2) during 10 minutes. 0.1 mmol of Cd(Ac)2·2H2O dissolved in 1 ml of N-methylformamide served as a cadmium precursor. For the obtention of the CdSe nanoplatelets, 200 μl of the 9 atomic layers thick CdSe nanoplatelets, 800 μl of hexane, 0.5 ml of N-methylformamide and 25 μl of the selenium precursors were stirred for 5 minutes in a flask. The as-obtained NPLs were capped with selenium and presented a total of 11 atomic layers. In order to remove the excess of selenium precursor, the NPLs in N-methylformamide were precipitated with ethanol by centrifugation and resuspended in N-methylformamide. Then 200 μl of the cadmium precursors was added to the NPLs and let react for 2 minutes. These NPLs were then 13 layers thick and were purified by centrifugation with ethanol and resuspended in N-methylformamide. They could be transferred back to hexane by adding hexane and oleic acid (or oleylamine) to the NPLs in N-methylformamide.
Fig. 1(a) and (b) show 9 and 13 atomic layers thick CdSe nanoplatelets, respectively. Their contrast in TEM is very homogenous on the entire surface, and they exhibit regular shape with no patches.
Ligand exchange on the CdSe nanoplatelets
Ligand exchange reactions were based on ref. 11. The precursor of HS− was prepared as follows. In a vial 0.1 mmol of NaHS was dissolved in 1 ml of N-methylformamide. For the obtention of a precursor of HO−, 0.1 mmol of KOH is dissolved in 1 ml of N-methylformamide in a vial. Then, in a flask, 200 μl of the CdSe NPLs of different thicknesses, 800 μl of hexane, 0.5 ml of N-methylformamide and 25 μl of the HS− (respectively HO−) precursors were stirred for 5 minutes. The NPLs were completely transferred from the non-polar phase to the polar due to the ligand exchange.Measurement of the band gaps of CdSe nanoplatelets
Fig. S1† shows the evolution of the absorption spectra with increasing NPL thickness. The band gap was determined at the maximum of the first absorption peak.Computational details
All calculations were carried out with the ab initio Crystal09 (ref. 30) code, which allows, through a standard Linear Combination of Atomic Orbitals (LCAO) approach, the self-consistent solution of both Hartree–Fock (HF) and Kohn–Sham (KS) equations, as well as the efficient use of hybrid functionals, using periodic boundary conditions.Following the results obtained in a previous work,28 our investigations were carried out using the global hybrid B3PW91 functional.31 For CdSe, we used a Gaussian-type orbital (GTO) double-zeta basis set, and replaced the core electrons with effective core potentials (ECPs) including scalar relativistic effects, as described in ref. 27. In more detail, the SBKJC small-core ECP32 was applied for Cd and the SBKJC large-core ECP32 with a basis set augmented by one d polarization function33 for Se. As a consequence, the following electrons were treated explicitly: 4s2 4p6 4d10 5s2 for Cd and 4s2 4p4 for Se. As previously described for CdSe,27 basis sets were modified by setting exponents inferior to 0.10 to 0.10. It is to note that although hybrid functionals can be used with plane-wave (PW) basis sets as well, calculations remain so far computationally much more expensive.34 On the other hand, when combined with Gaussian-type basis sets, hybrids can efficiently be applied to periodic systems,35,36 even to very large ones like the nanoplatelets studied here.37
To simplify calculations, HCOO− served as a model for fatty acids, keeping in mind that it is only through the carboxylate functional group that fatty acids bind to the surface, and the alkyl chain has no influence on the changes induced in the surface electronic states upon adsorption of the ligand. All-electron GTO basis sets taken from ref. 38 were applied for the modeling of the C, O and H atoms of the adsorbed species with the following contractions: (9s, 3p, 1d) → [3s, 2p, 1d] for C atoms, (14s, 6p, 1d) → [4s, 3p, 1d] for O atoms, (7s, 1p) → [3s, 1p] for H atoms, and (14s, 8p, 1d) → [5s, 4p, 1d] contraction for S atoms.39
An extra-large DFT integration grid, consisting of 75 radial and 974 angular points, was applied in order to provide accurate results for calculations. The following truncation criteria (ITOL parameters)30 were set for the accuracy of the Coulomb and exchange series: 10−7 as the overlap threshold for Coulomb integrals, 10−7 as the penetration threshold for Coulomb integrals, 10−7 as the overlap threshold for HF exchange integrals, and 10−9 and 10−20 as the pseudo-overlap for HF exchange series. A Monkhorst–Pack shrinking factor40 of 6 has been applied, which corresponds to 20 k points in the irreducible Brillouin zone (IBZ) of the stabilized zinc blende (100) surfaces.
For the calculations of surface properties, we adopted a slab model: a slice of material with a thickness of several atomic layers, terminated by two free surfaces, for which two dimensional periodic boundary conditions were imposed. Only atoms were allowed to relax during geometry optimization in the stabilised CdSe zinc blende slabs. All of the slabs studied here are non-stoichiometric, and are terminated by Cd atoms on both basal planes like the CdSe zinc blende nanoplatelets previously synthesized by Ithurria et al.5 Two adsorption modes of formiate and one adsorption mode of hydroxide and sulfide ions on CdSe (100) surface were considered. In the following, the studied systems are abbreviated as CdSe·HCOO, CdSe·OH and CdSe·SH, respectively.
In general, the adsorption energy of an X ligand on the surface of Y can be calculated as:
| Eads = Etot(Y·X) − Etot(X) − Etot(Y) | (1) |
Instead, the zinc blende (100) surface is a polar, unstable Tasker type 3 surface, that is characterized by a divergent surface energy originating from the permanent dipole momenta perpendicular to its surface.41 The third term (the energy of the clean slab) in eqn (1) is therefore approximated as the total energy of a part of the bulk crystal which contains a number of atoms equal to that of the slab.
Furthermore, since the Cd-terminated (100) slab is nonstoichiometric, the first term of eqn (1) is replaced by the average of the total energies of a slab terminated by Cd atoms and stabilized with anionic ligands on both basal planes and that of a CdSe zinc blende slab terminated by Se atoms and stabilized by Na+ ions on its basal planes. This approach is similar to the one proposed for the calculations of non-stoichiometric perovskite slabs by Evarestov et al.42 Next, the total energy of both the anion and the Na+ should be subtracted from this term.
To sum up, in this paper, the adsorption energies of HCOO−, OH− and SH− ligands on the CdSe zinc blende (100) surface were thus calculated according to the following equation:
| Eads = (Etot(CdSe·X) + Etot(CdSe·Na))/2 − Etot(X−) − Etot(Na+) − nEtot(CdSebulk) | (2) |
Results and discussion
CdSe zinc blende (100) surface
Under atmospheric conditions, two stable phases are found for CdSe: the hexagonal wurtzite structure and the face-centered cubic zinc blende structure,10 the latter belonging to the Fm3m space group. The zinc blende (100) surface consists of alternately charged planes,41 as also shown in Fig. 1. In this structure, the dipole moments originating from the two dangling bonds of the under-coordinated Cd atoms in the uppermost layer of the (100) basal planes sum up as a dipole moment perpendicular to the (100) surface (as shown on Fig. 1), which results in a divergent surface energy. In order to stabilize this kind of surfaces, a substantial surface reconstruction or the adsorption of additional species is needed.Because of their instability, polar semiconductor surfaces are less investigated than cleavage surfaces like the wurtzite (10−10) and (11−20) of CdSe.21,22,43 In theoretical studies, polar CdSe surfaces have been passivated by phosphines,44,45 amines,44,46 thiols,44 and fatty acids.47,48 These organic ligands are commonly used during and after the synthesis of CdSe quantum dots.5,49,50 Atomic reconstructions of the polar zinc blende (111) and (001) surfaces of CdSe have also been investigated,51 the latter surface being equivalent to the (100) surface of our interest.
Carefully chosen ligands can influence the shape of the nanocrystal.14,52,53 The CdSe zinc blende crystal structure has an isotropic unit cell structure, which facilitates the growth of isotropic structures like cubes. In order to produce quasi-two-dimensional (2D) zinc blende nanocrystals, one needs to suppress the high reactivity of polar surfaces.47,54 As all the above mentioned ligands link to cations, a thin quasi-2D (100) nanocrystal whose synthesis is based on the passivation of polar surfaces should be terminated by Cd atoms on both (100) basal planes, as in previously studied CdSe zinc blende nanoplatelets.17,47 However, this results in a non-stoichiometric structure and excess positive charges due to the additional Cd plane, which should be compensated by negative charges. Li et al.47 proposed a structure in which onefold negatively charged ligands are adsorbed on both basal planes of a nanocrystal. Indeed, in this case, in a purely ionic picture, the additional +2 charge per unit cell due to the excess of Cd planes is neutralized by the 2·(−1) charge of the ligands on the two basal planes, as long as each surface Cd is bound to a ligand. The appropriately chosen ligands also serve as a model of different capping agents of the synthesized nanoplatelets, which influence their charge transfer properties,11 therefore this way of stabilizing the zinc blende (100) surface leads to a model that is better adapted to simulate the synthesized nanoplatelets than the surface atomic reconstructions. In the present work, calculations were performed only on slabs with selected thicknesses: 5, 9 and 13 atomic layers.
Geometrical investigation
Fig. 2 represents the optimized geometries of five layers thick slabs stabilized with different ligands. With the chosen orientation, the ligands on the two basal planes are turned by 90° with respect to each other, following the relative positions of tetracoordinated Cd and Se atoms in the bulk crystal. The optimized geometrical parameters are reported in Table 1. It is to note that the interatomic distances characterizing the optimized adsorption geometries of the ligands on the CdSe zinc blende (100) surface do not change with slab thickness. | ||
| Fig. 2 Optimized structures of stabilized five layer thick slabs: the CdSe·HCOO system in the (a) bidentate and (b) monodentate adsorption mode of the formiate ligand, and those of the (c) CdSe·SH and (d) CdSe·OH systems. Cd, Se, O, C, H and S atoms are presented as beige, orange, red, grey, white and yellow spheres, respectively. | ||
| CdSe·HCOO | CdSe·SH | CdSe·OH | |||
|---|---|---|---|---|---|
| d(C–O1) | 2.43 | d(Cd1a–S1) | 2.60 | d(Cd1a–O) | 2.34 |
| d(C–O2) | 2.37 | d(Cd1b–S2) | 2.61 | d(Cd1b–O) | 2.34 |
| d(C–O1′) | 2.27 | d(S1–H1) | 1.37 | d(O1–H1) | 0.96 |
| d(Cd1a–C) | 2.75 | d(S1–H2) | 3.58 | d(O1–H2) | 3.62 |
| d(Cd1b–C) | 2.75 | d(S2–H2) | 1.37 | d(O2–H2) | 0.96 |
| d(C–H) | 1.10 | d(S2–H1) | 2.75 | d(O2–H1) | 3.62 |
In the present study, the mono- and bidentate adsorption geometries of the formiate ion (Fig. S2(a) and (b),† respectively) were considered on the CdSe zinc blende (100) surface. As mentioned above, the excess +2 charge per unit cell (see Fig. 2) of the additional Cd layer is compensated in these cases, as one HCOO− anion is attached to each Cd atom on both basal planes, which means that 2·(−1) = −2 charge is introduced upon ligand adsorption. A test optimization on five layer slabs showed that the two optimized structures actually converge on the same minimum. Starting either with a mono- or with a bidentate adsorption mode resulted in a geometry in which the formiate group is tilted in a way that one of its O atoms (O1) forms a bridge between two neighboring Cd atoms and the other O atom (O2) stays connected to one single Cd atom, resulting in pentacoordinated Cd atoms, as shown on Fig. 2(a) and (b). As a consequence, for further investigations on thicker slabs, only the bidentate adsorption mode was considered as initial geometry.
Also, in the case of SH− ligands, the initial configuration was chosen to be a bridging one (see Fig. 2(c)). This structure can be regarded as a result of a surface treatment of the CdSe zinc blende (100) surface with sulfide ions that occupy the same positions as selenide ions in a bulk zinc blende CdSe crystal.
The tetrahedral coordination of the surface Cd atoms is thus ensured. Each surface Cd atom is linked to 2 Se and 2 S atoms, and each S atom is linked to two Cd atoms and one H atom. In this arrangement, the negative charges introduced by the so formed S2− layers (−2 per unit cell) are compensated by H+ counter ions (2·(+1) per unit cell), similarly as described above for CdSe·HCOO. As a result of the optimization, S atoms are displaced closer to H atoms of the neighbouring bridges, possibly forming a network of S–H hydrogen bonds. Regarding the distance between S and H atoms of the neighbouring bridges, alternating rows are formed on the (100) basal planes, characterized by small but remarkable differences in the relative positions of the atoms, as reported in Table 1.
Finally, the initial structure for the OH− stabilized CdSe zinc blende (100) slabs was constructed based on the relaxed CdSe·SH geometry: the S atoms of the relaxed SH− stabilized slabs were simply replaced by O atoms. Upon optimization, the O atoms got closer to the surface than S atoms (Fig. 2d), and instead of alternating rows, all optimized O–H distances are equal (Table 1).
Adsorption energies
The adsorption energies of the studied ligands on slabs of different thickness have been calculated based on eqn (2). Bifunctional linkers that are used to attach CdSe quantum dots to wide band gap semiconductors such as ZnO and TiO2 are typically mercaptoalcanoic acids, e.g. mercaptopropionic acid (MPA, HS–CH2–CH2–COOH).55–57 These molecules link to the QD with their sulfide functional group, and their carboxylate group bounds to the wide band gap oxide. This is in line with the reported average adsorption energies in Fig. 3: the absolute value of the obtained adsorption energy of the SH− ligand on the CdSe zinc blende (100) surface is higher than that of the HCOO− ligand. One can also see in Fig. 3 that according to the established theoretical model, the OH− ligand is attached even more strongly to the CdSe zinc blende (100) surface than the SH− ligand. In contrary to this result, as mentioned above, Nag et al. previously observed that contrary to SH− ligands, OH− ligands cannot be completely exchanged with the fatty acids used as capping agents during the synthesis of colloidal CdSe quantum dots,11 from which one could conclude that the adsorption energy of SH− on CdSe QDs is superior to that of OH−. However, it should be noted that the spherical quantum dots used in their study expose several different surfaces58 to which the adhesion of ligands can be different.59,60 Most importantly, one can remark from Fig. 3 that the adsorption energy of the studied ligands is practically independent of the slab thickness, confirming the convergence of results on the overall of the chosen thicknesses. | ||
| Fig. 3 Adsorption energies of HCOO−, OH− and SH− ligands on the CdSe zinc blende (100) surface (in kcal mol−1) calculated according to eqn (2) for 5; 9 and 13 atomic layers thick slabs. | ||
Electronic properties
Mulliken charge analysis performed for 5 and 13 atomic layers thick CdSe zinc blende slabs stabilized with different ligands on their (100) surfaces is reported in Table S1.† One can conclude that the difference between the charges of corresponding atoms in the uppermost layers of the slabs is negligible in case of 5 and 13 layers thick slabs with the same ligands, which indicates that increasing the thickness of these CdSe nanoplatelets does not significantly change the electronic properties of the zinc blende (100)–ligand interface, in line with the practically constant adsorption energies of the different ligands on this surface as a function of slab thickness (see before). In case of 13 layers thick slabs, regarding the evolution the absolute values of the atomic charges of Cd and Se from the (100) surface towards the innermost layers in the [100] direction, they both converge to a value of around 0.42–0.43|e−| (where |e−| is the elementary electron charge), close to the previously obtained 0.424|e−| value for bulk CdSe.28 This shows that the ions in the innermost layers of a 13 layers thick CdSe zinc blende slab already possess bulk-like charges.The total charge of the adsorbed hydroxide ligand (−0.644|e−|) is close to that of the adsorbed formiate ligand (−0.663|e−|). The relatively high atomic charges of the surface Cd (0.880–0.882|e−|) and the O atoms involved in the binding of the OH− (−0.950|e−|) and HCOO− ligands (qO1 = −0.485|e−| and qO2 = −0.610|e−|) and the low bond overlap populations between them (bCd–O = 0.150|e−| for CdSe·OH, bCd–O1 = 0.038|e−|, bCd–O2 = 0.130|e−| and bCd–O1′ = 0.018|e−| for CdSe·HCOO) suggest a significantly ionic character of the Cd–O bonding. On the other hand, in case of the surfaces stabilized with SH− ligands, both S and corresponding Cd atoms exhibit much weaker charges (qCd = 0.341–0.348|e−|, qS1 = −0.098|e−| and qS2 = −0.116|e−|), and one can observe a significantly higher bond overlap population between the surface Cd and S atoms (bCd–S = 0.452|e−|), suggesting the strong covalent character of the Cd–S bond.
Indeed, from the computed charges it seems that a pronouncedly effective charge transfer mechanism from the SH− to the CdSe slab surface takes place, the SH− moiety being only very marginally negatively charged when adsorbed on the surface (of the order of −0.1|e−|). Furthermore, as a consequence to the not fully symmetric arrangement of the two SH− ligands, the H1 and S2 atoms, that are closer to each other are more negatively charged than the H2 and S1 atoms. This pattern of charges induced by ligands with alternate configurations is observable until the third layer in the CdSe·SH slabs.
In more detail, the Mulliken charge analysis of the individual Cd, O and S atomic orbitals of the CdSe–ligand interface is reported in Table S2† for 5 and in Table 2 for 13 layers thick slabs. One can remark that in case of the CdSe·SH system, the 6sp orbital of surface Cd atoms is filled with more electrons (0.835 or 0.853) than in the CdSe·OH (0.367) and CdSe·HCOO system (0.250), while the other corresponding orbitals of these systems are filled with nearly the same amount of electrons. This indicates that the above mentioned charge transfer from the ligands towards the CdSe zinc blende slab is directed towards the 6 sp orbitals of the surface Cd atoms. The total and orbital-projected density of states (DOS) together with the corresponding band structures are shown in Fig. 4. The reported figures correspond to 13 layers thick slabs, in case of which the atomic charges of the innermost layers are already converged to the bulk value, as previously discussed in Table S1.† The overall flat structure of the bands with the nonetheless observable energy dispersion shows the compounds studied here have a partially ionic, partially covalent character. It is also important to note that none of the ligands introduce electronic states in the band gap.
| CdSe·HCOO | |||||||
| Cd1 | 4sp | 4d | 5sp | 5d | 6sp | 6d | 7sp |
| 4.171 | 5.820 | 0.908 | 3.224 | 0.250 | 0.998 | 3.750 | |
| O1 | 1sp | 2sp | 3sp | 3d | 4sp | ||
| 1.998 | 2.715 | 2.660 | 0.024 | 1.087 | |||
| O2 | 1sp | 2sp | 3sp | 3d | 4sp | ||
| 1.999 | 2.715 | 2.698 | 0.020 | 1.180 | |||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| CdSe·SH | |||||||
| Cd1a | 4sp | 4d | 5sp | 5d | 6sp | 6d | 7sp |
| 4.181 | 5.797 | 0.851 | 3.204 | 0.853 | 1.031 | 3.742 | |
| Cd1b | 4sp | 4d | 5sp | 5d | 6sp | 6d | 7sp |
| 4.181 | 5.797 | 0.862 | 3.203 | 0.835 | 1.032 | 3.742 | |
| S1 | 1s | 2s + 2p | 3s + 3p | 3d | 4s + 4p | 5s + 5p | |
| 2.010 | 4.759 | 3.313 | 0.049 | 5.294 | 0.677 | ||
| S2 | 1s | 2s + 2p | 3s + 3p | 3d | 4s + 4p | 5s + 5p | |
| 2.010 | 4.759 | 3.313 | 0.049 | 5.292 | 0.695 | ||
|
|||||||
| CdSe·OH | |||||||
| Cd1 | 4sp | 4d | 5sp | 5d | 6sp | 6d | 7sp |
| 4.171 | 5.815 | 0.826 | 3.223 | 0.367 | 0.993 | 3.750 | |
| O | 1sp | 2sp | 3sp | 3d | 4sp | ||
| 1.998 | 2.682 | 2.737 | 0.016 | 1.516 | |||
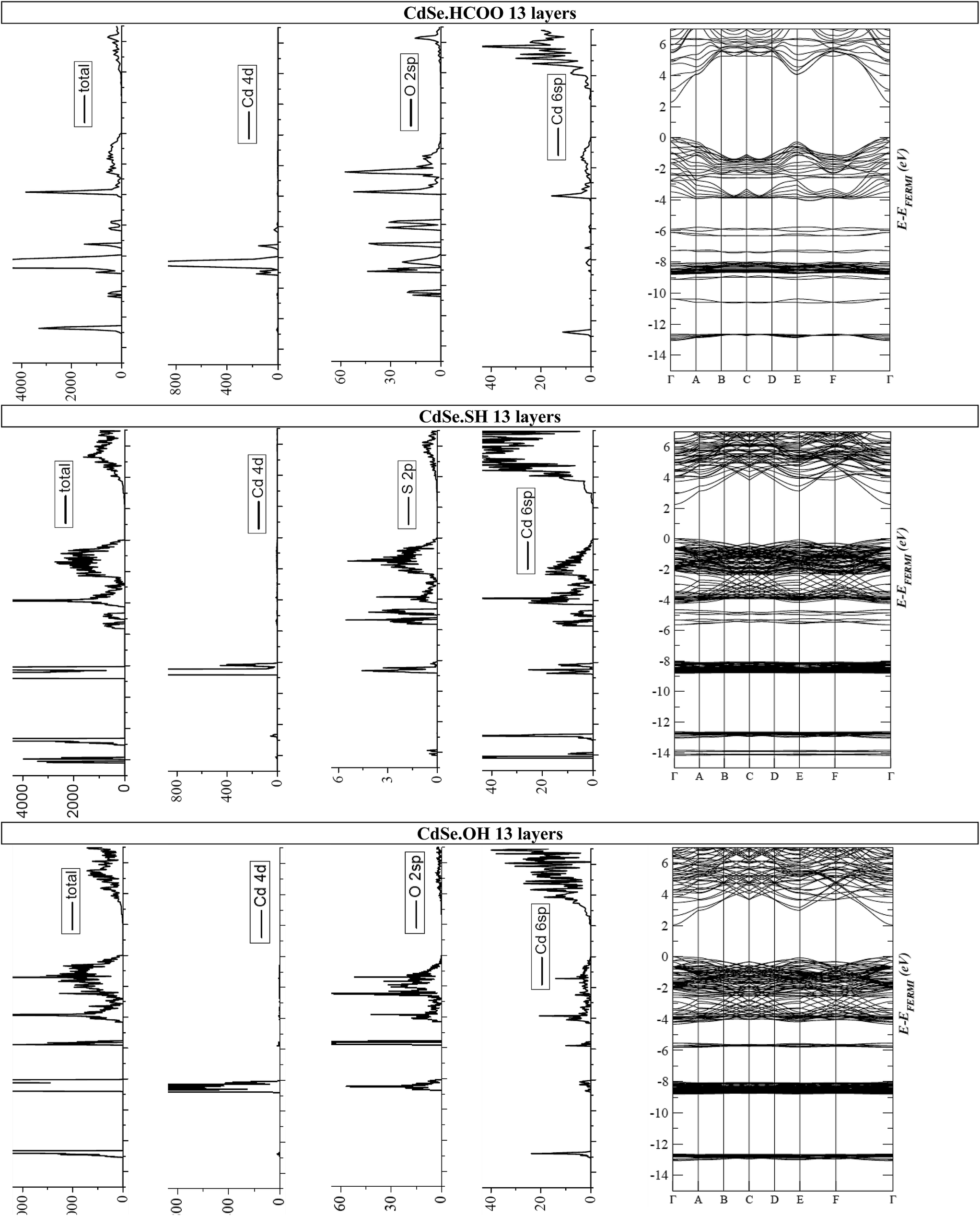 | ||
| Fig. 4 Total and orbital-projected density of states of the S and O atoms of the ligands and of the Cd atoms of the (100) surface and band structures of the (a) CdSe·HCOO, (b) CdSe·SH and (c) CdSe·OH systems. Fermi level was set at 0 eV. | ||
As regards the Cd atoms of the (100) surfaces of the systems studied here, their 4d orbitals mainly contribute to the valence band of the stabilized slabs. In the CdSe·HCOO and CdSe·OH, the 2sp of the O atoms also contribute to the DOS in the valence band. The overlap between these two orbitals suggests a partially covalent character of the Cd–O bond on the (100) surfaces of the slabs. Supposing an analogy with the CdSe·OH system, the S 3p orbital should overlap with the Cd 4d in the CdSe·SH. However, it does not contribute to the total DOS, only the S 2p core orbital, which, on the other hand, overlaps with Cd 4d, showing the covalent character of the Cd–S bond as well. Also, in case of the Cd 6sp orbital, which is more negatively charged in the CdSe·SH than in the CdSe·OH and CdSe·HCOO system, the peak at around −14 eV is significantly more intense than in the other two slabs. These observations support again the above described charge transfer from the SH− ligand to the Cd terminated CdSe zinc blende (100) surface.
The computed band gaps as a function of slab thickness together with the experimental values are listed in Table 3 and represented in Fig. 5. Taken all together, the computed data are in very good agreement with the experimental ones both in terms of trend and absolute value.
| n | 5 | 7 | 9 | 11 | 13 | |
|---|---|---|---|---|---|---|
| HCOO− | Computed | 3.31 | nd | 2.57 | nd | 2.29 |
| Experimental | 3.13 | 2.68 | 2.42 | 2.25 | 2.16 | |
| SH− | Computed | 3.00 | nd | 2.48 | nd | 2.25 |
| Experimental | nd | 2.46 | 2.29 | 2.15 | nd | |
| OH− | Computed | 2.50 | nd | 2.17 | nd | 2.05 |
| Experimental | nd | 2.50 | 2.39 | 2.21 | nd |
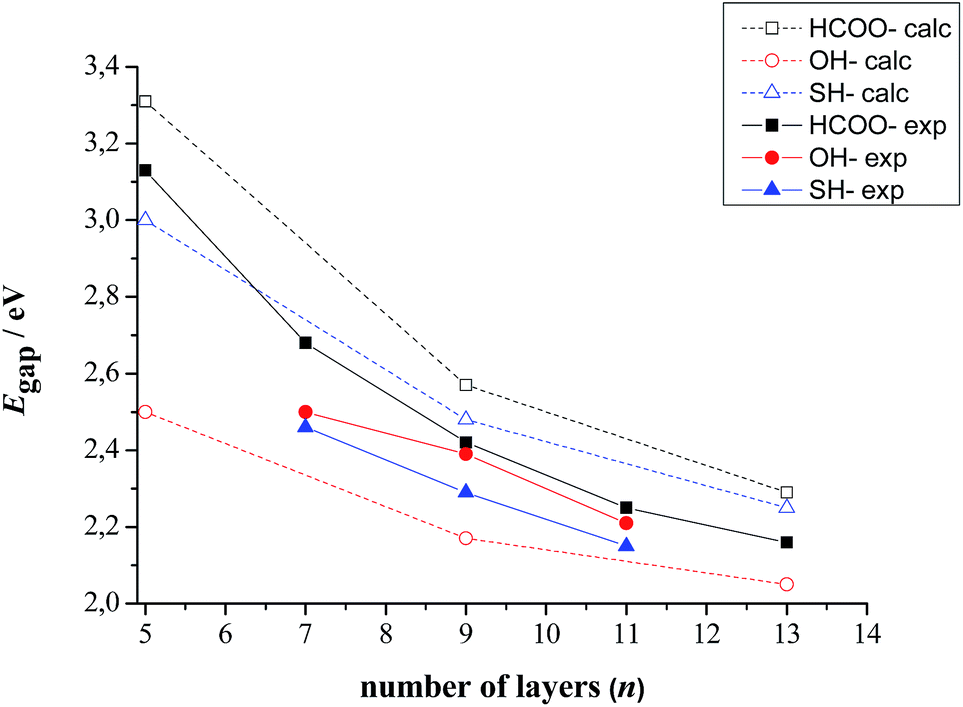 | ||
| Fig. 5 Calculated and experimental band gaps as a function of slab thickness for CdSe zinc blende slabs stabilized with HCOO−, SH− and OH− ligands on their (100) basal planes. Note that experimentally in the case of OH− ligand exchange is not complete.11 | ||
In more detail, the results show that the gap is slightly overestimated for the HCOO− and SH− ligands, while the reverse holds for OH− ligands. It can also be noted that the experimental Egap(CdSe·OH) > Egap(CdSe·SH) order is inversed in case of the computed results. It should be kept in mind however, that as it has previously been shown by FTIR spectroscopy,11 the ligand exchange reaction is not complete for OH− ligands, contrary to SH− ligands. Therefore, the synthesized, OH− passivated CdSe zinc blende 2D nanocrystals are, in reality, covered by a mix of hydroxide ligands and, in an inferior proportion, fatty acids, which can indeed increase their band gap compared to a purely OH− capped nanoplatelet like the established CdSe·OH model.
Conclusions
In this study, a comprehensive density functional theory investigation has been performed on nonstoichiometric CdSe zinc blende nanoplatelets of various thickness, stabilized by different ligands (HCOO−, SH− and OH−) on their polar (100) surface. A theoretical model has been established for these hybrid ligand–nanocrystal systems. The global hybrid B3PW91 functional was used for all calculations, combined with the SBKJC small- and large-core effective pseudopotentials for Cd and Se, respectively, and all-electron basis sets for H, C, S and O atoms. The relaxation parameters of the ligands adsorbed on the (100) surface of the nanoplatelets have been calculated, along with their adsorption energies. We found that these parameters do not change with the thickness of the nanoplatelets, confirming the convergence of these properties on the overall of the chosen thicknesses. Regarding the electronic properties, the band gaps, band structures and orbital-projected density of states of the stabilized nanoplatelets have been calculated, along with a detailed Mulliken analysis of the atomic and orbital charges. The latter revealed a major electron transfer from the SH− ligands towards the surface of the nanocrystals, in line with what can be inferred from the density of states. CdSe zinc blende nanoplatelets of various thicknesses, stabilized by fatty acids, SH− and OH− ligands have also been synthesized, and their band gaps have been measured by absorption spectroscopy. A nice agreement is found between the experimental and calculated band gaps, especially concerning the evolution of the band gaps with the thickness of the nanoplatelets. We can thus conclude that the chosen theoretical model and computational protocol together can serve as a powerful tool for the qualitative and quantitative description of the geometrical and electronic properties of CdSe nanoplatelets, which can potentially be applied in various advanced applications.Acknowledgements
This work has been supported by the Region Ile-de-France in the framework of DIM Nano-K (Nanodot-PV project). The authors acknowledge IDRIS for providing computational resources. Paris Sciences et Lettres (PSL) Research University is acknowledged for financial support (QuanDox project).Notes and references
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510 CrossRef CAS.
- A. G. Midgett, J. M. Luther, J. T. Stewart, D. K. Smith, L. A. Padilha, V. I. Klimov, A. J. Nozik and M. C. Beard, Nano Lett., 2013, 13, 3078 CrossRef CAS PubMed.
- J. H. Bang and P. V Kamat, ACS Nano, 2009, 3, 1467 CrossRef CAS PubMed.
- A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V Kamat, J. Am. Chem. Soc., 2008, 130, 4007 CrossRef CAS PubMed.
- S. Ithurria, M. D. Tessier, B. Mahler, R. P. S. M. Lobo, B. Dubertret and A. L. Efros, Nat. Mater., 2011, 10, 936 CrossRef CAS PubMed.
- A. W. Achtstein, A. Schliwa, A. Prudnikau, M. Hardzei, M. V Artemyev, C. Thomsen and U. Woggon, Nano Lett., 2012, 12, 3151 CrossRef CAS PubMed.
- A. E. Kuznetsov, D. Balamurugan, S. S. Skourtis and D. N. Beratan, J. Phys. Chem. C, 2012, 116, 6817 CAS.
- S. Jain, S. N. Sharma and M. Kumar, Phys. E, 2011, 44, 555 CrossRef CAS.
- A. Franceschetti and Y. Zhang, Phys. Rev. Lett., 2008, 100, 136805 CrossRef PubMed.
- O. Zakharov, A. Rubio and M. L. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4926 CrossRef CAS.
- A. Nag, M. V Kovalenko, J. Lee, W. Liu, B. Spokoyny and D. V Talapin, J. Am. Chem. Soc., 2011, 133, 10612 CrossRef CAS PubMed.
- J. L. Baker, A. Widmer-Cooper, M. F. Toney, P. L. Geissler and A. P. Alivisatos, Nano Lett., 2010, 10, 195 CrossRef CAS PubMed.
- J. S. Jie, W. J. Zhang, Y. Jiang and S. T. Lee, Appl. Phys. Lett., 2006, 89, 133118 CrossRef.
- L. Manna, D. J. Milliron, A. Meisel, E. C. Scher and A. P. Alivisatos, Nat. Mater., 2003, 2, 382 CrossRef CAS PubMed.
- J. S. Son, X.-D. Wen, J. Joo, J. Chae, S.-I. Baek, K. Park, J. H. Kim, K. An, J. H. Yu, S. G. Kwon, S.-H. Choi, Z. Wang, Y.-W. Kim, Y. Kuk, R. Hoffmann and T. Hyeon, Angew. Chem., Int. Ed., 2009, 48, 6861 CrossRef CAS PubMed.
- (a) S. Ithurria and B. Dubertret, J. Am. Chem. Soc., 2008, 130, 16504 CrossRef CAS PubMed; (b) E. Lhuillier, S. Pedetti, S. Ithurria, H. Heuclin, B. Nadal, A. Robin, G. Patriarche, N. Lequeux and B. Dubertret, ACS Nano, 2014, 8, 3813 CrossRef CAS PubMed.
- M. D. Tessier, C. Javaux, I. Maksimovic, V. Loriette and B. Dubertret, ACS Nano, 2012, 6, 6751 CrossRef CAS PubMed.
- L. T. Kunneman, M. D. Tessier, H. Heuclin, B. Dubertret, Y. V Aulin, F. C. Grozema, J. M. Schins and L. D. A. Siebbeles, J. Phys. Chem. Lett., 2013, 4, 3574 CrossRef CAS.
- C. Bouet, B. Mahler, B. Nadal, B. Abecassis, M. D. Tessier, S. Ithurria, X. Xu and B. Dubertret, Chem. Mater., 2013, 25, 639 CrossRef CAS.
- F. Xu, W. Zhou and A. Navrotsky, J. Mater. Res., 2011, 26, 720 CrossRef CAS.
- Y. R. Wang and C. B. Duke, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 6417 CrossRef CAS.
- I. Csik, S. P. Russo and P. Mulvaney, Chem. Phys. Lett., 2005, 414, 322 CrossRef CAS.
- S. Sarkar, S. Pal, P. Sarkar, A. L. Rosa and T. Frauenheim, J. Chem. Theory Comput., 2011, 7, 2262 CrossRef CAS.
- A. Puzder, A. J. Williamson, N. Zaitseva, G. Galli, L. Manna and A. P. Alivisatos, Nano Lett., 2004, 4, 2361 CrossRef CAS.
- T. Le Bahers, F. Labat, T. Pauporté, P. P. Lainé and I. Ciofini, J. Am. Chem. Soc., 2011, 133, 8005 CrossRef CAS PubMed.
- T. Le Bahers, F. Labat, T. Pauporté and I. Ciofini, Phys. Chem. Chem. Phys., 2010, 12, 14710 RSC.
- H. Xiao, J. Tahir-Kheli and W. A. Goddard, J. Phys. Chem. Lett., 2011, 2, 212 CrossRef CAS.
- A. Szemjonov, T. Pauporté, I. Ciofini and F. Labat, Phys. Chem. Chem. Phys., 2014, 16, 23251 RSC.
- S. Ithurria and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 18585 CrossRef CAS PubMed.
- (a) R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571 CAS; (b) R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D'Arco, and M. Llunell, CRYSTAL09 User's Manual, University of Torino, Torino, 2013 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 7, 5648 CrossRef.
- W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Can. J. Chem., 1992, 70, 612 CrossRef CAS.
- N. P. Labello, A. M. Ferreira and H. a. Kurtz, J. Comput. Chem., 2005, 26, 1464 CrossRef CAS PubMed.
- M. Marsman, J. Paier, a. Stroppa and G. Kresse, J. Phys. Condens. Matter, 2008, 20, 064201 CrossRef CAS PubMed.
- M. D. Towler, A. Zupan and M. Causa, Comput. Phys. Commun., 1996, 98, 181 CrossRef CAS.
- F. Corà, M. Alfredsson, G. Mallia, D. S. Middlemiss, W. C. Mackrodt, R. Dovesi and R. Orlando, in Structure and Bonding, Springer-Verlag Berlin Heidelberg, 2004, vol. 113, p. 171 Search PubMed.
- R. Orlando, M. Delle Piane, I. J. Bush, P. Ugliengo, M. Ferrabone and R. Dovesi, J. Comput. Chem., 2012, 33, 2276 CrossRef CAS PubMed.
- F. Labat, I. Ciofini and C. Adamo, J. Chem. Phys., 2009, 131, 044708 CrossRef PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- P. W. Tasker, J. Phys. C: Solid State Phys., 1979, 12, 4977 CrossRef CAS.
- R. a. Evarestov, E. a. Kotomin, D. Fuks, J. Felsteiner and J. Maier, Appl. Surf. Sci., 2004, 238, 457 CrossRef CAS.
- B. Siemens, C. Domke, P. Ebert and K. Urban, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 12321 CrossRef CAS.
- P. Schapotschnikow, B. Hommersom and T. J. H. Vlugt, J. Phys. Chem. C, 2009, 113, 12690 CAS.
- E. Rabani, J. Chem. Phys., 2001, 115, 1493 CrossRef CAS.
- L. Manna, L. W. Wang, R. Cingolani and a. P. Alivisatos, J. Phys. Chem. B, 2005, 109, 6183 CrossRef CAS PubMed.
- Z. Li and X. Peng, J. Am. Chem. Soc., 2011, 133, 6578 CrossRef CAS PubMed.
- L. Liu, Z. Zhuang, T. Xie, Y.-G. Wang, J. Li, Q. Peng and Y. Li, J. Am. Chem. Soc., 2009, 131, 16423 CrossRef CAS PubMed.
- K. Yu, S. Singh, N. Patrito and V. Chu, Langmuir, 2004, 20, 11161 CrossRef CAS PubMed.
- J. K. Cooper, A. M. Franco, S. Gul, C. Corrado and J. Z. Zhang, Langmuir, 2011, 27, 8486 CrossRef CAS PubMed.
- L. Zhu, K. L. Yao, Z. L. Liu and Y. B. Li, J. Phys.: Condens. Matter, 2009, 21, 095001 CrossRef CAS PubMed.
- D. V. Talapin, A. L. Rogach, A. Kornowski, M. Haase and H. Weller, Nano Lett., 2001, 1, 207 CrossRef CAS.
- R. Benchamekh, N. a. Gippius, J. Even, M. O. Nestoklon, J.-M. Jancu, S. Ithurria, B. Dubertret, A. L. Efros and P. Voisin, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 035307 CrossRef.
- R. Benchamekh, J. Even, J. Jancu, M. Nestoklon, S. Ithurria, B. Dubertret and P. Voisin in Nanosctructures: Physics and Technology, Nizhny Novgorod, Russia, 2012, p. 56 Search PubMed.
- I. Mora-Seró, S. Giménez, T. Moehl, F. Fabregat-Santiago, T. Lana-Villareal, R. Gómez and J. Bisquert, Nanotechnology, 2008, 19, 424007 CrossRef PubMed.
- I. Robel, V. Subramanian, M. Kuno and P. V Kamat, J. Am. Chem. Soc., 2006, 128, 2385 CrossRef CAS PubMed.
- R. S. Dibbell and D. F. Watson, J. Phys. Chem. C, 2009, 113, 3139 CAS.
- Y. A. Yang, H. Wu, K. R. Williams and Y. C. Cao, Angew. Chem., Int. Ed., 2005, 44, 6712 CrossRef CAS PubMed.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 1389 CrossRef CAS.
- J. S. Owen, J. Park, P.-E. Trudeau and A. P. Alivisatos, J. Am. Chem. Soc., 2008, 130, 12279 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Evolution of the absorption spectra with the CdSe NPL thickness, initial geometries of CdSe·HCOO systems with bidentate and monodentate adsorption modes, computed Mulliken atomic charges for 5 and 13 atomic layers thick CdSe·HCOO, CdSe·OH and CdSe·SH systems, number of electrons on Cd, S and O atomic orbitals 5 atomic layers thick CdSe·HCOO, CdSe·OH and CdSe·SH systems. See DOI: 10.1039/c4ra10082d |
| This journal is © The Royal Society of Chemistry 2014 |