DOI:
10.1039/C4RA09989C
(Paper)
RSC Adv., 2014,
4, 57591-57599
Preparation, characterization and analytical application of an electrochemical laccase biosensor towards low level determination of isoprenaline in human serum samples
Received
8th September 2014
, Accepted 22nd October 2014
First published on 23rd October 2014
Abstract
A novel electrochemical biosensor has been developed based on the immobilization of multiwalled carbon nanotubes (MWCNT) on to a glassy carbon electrode (GCE) and subsequent casting of silica sol–gel (SiSG) entrapped laccase (Lac) enzyme on to the MWCNT/GCE. The catalytic activity of laccase biosensor was found to be good enough for sensitive determination of isoprenaline (ISP) with the aid of voltammetric techniques and we have also demonstrated the detailed electrochemical redox mechanism of ISP. From the effect of the pH, we have optimized the optimum pH as 6.5, and from effect of scan rate we have evaluated the kinetic parameters, heterogeneous rate constant, charge transfer coefficient and diffusion coefficient values. Furthermore the limit of detection (LOD) and limit of quantification (LOQ) values were found to be 1.8 × 10−7 M and 6.0 × 10−7 M, respectively. The simultaneous determination of ISP in the presence of uric acid (UA) and ascorbic acid (AA) was successfully carried out. The surface nature of the biosensor was characterized by using electrochemical impedance spectroscopy. Finally the validation of the proposed method was verified by the recovery of injection (ISP) in serum samples and their recoveries were found to be in a satisfactory range. The proposed method was found to have good repeatability, reproducibility and stability with low relative standard deviation (RSD) values.
1. Introduction
Isoprenaline (ISP) (4-[1-hydroxy-2-(isopropylamino)ethyl]benzene-1,2-diol), also known as isoproterenol, is a catecholamine drug, which has been used for bradycardia or heart block. ISP activates the β1-receptors on the heart and shows positive chronotropic, dromotropic and inotropic effects.1–3 It was also used for the treatment of primary pulmonary hypertension, status asthmaticus and bronchial asthama.4–7 ISP can give relaxation from all varieties of smooth muscle when the tone is high.8 The excessive use of ISP, however, causes heart failure and arrhythmias. In 1963, the UK, Australia, New Zealand and three other countries encountered a series of deaths, which were associated with repeated and excessive use of ISP inhalation.9,10 Therefore, there was an essentiality for the development of new sensors for the quantitative determination of ISP levels in human blood samples.
Ascorbic acid (AA) and uric acid (UA) are the most important electroactive biological compounds present in the human body, which play a potential role in the metabolic system and show similar electrochemical behaviour as catecholamines. Ascorbic acid (AA) is important in the health care of humans and is especially essential to the skin, connective tissues and immune system. Uric acid (UA) is the final oxidation product of urine metabolism and is excreted in the urine.11 At conventional electrodes the determination of catecholamines in the presence of UA and AA is difficult owing to overlap of voltammetric responses.12,13 Adsorption of AA products onto conventional electrodes surface causes surface fouling, poor selectivity and poor reproducibility.14 Hence it is a challenging task for the simultaneous determination of ISP, AA and UA. So far a very few mediators have been reported towards the simultaneous determination of ISP, which includes conducting material,15 carbon nanotube paste electrode,7,16 metal complexes and graphene modified electrodes.3
Since, from the discovery of CNTs in 1991 by Sumio Iijima, they have emerged as a novel class of nanomaterials having applications in chemistry and physics. Due to the good structural, mechanical, electrical and physical properties of CNTs, they have been employed for the preparation of CNT-modified electrodes.17 These CNTs enhance the voltammetric peak height, which facilitates the sensitive determination in analytical sensing, while also reducing the overpotential of the system with no surface fouling.18,19
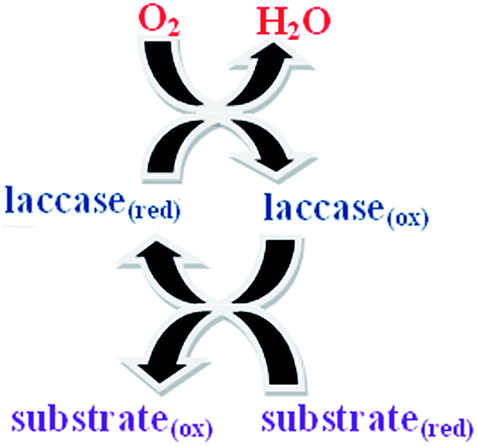
Laccases belong to a group of polyphenol oxidases containing copper atoms in their catalytic center, which are denoted as multi copper oxidases. These enzymes are widely described in plants, fungi (ascomycetes and basidiomycetes) and microorganisms, where they are presumably involved in lignin synthesis and degradation processes. Additionally, laccases can play a role in fungal virulence by the detection from phytoalexins and tannins. The catalytic center of the laccase cluster has four copper atoms and undergoes oxidation by oxygen and is brought back to its reduced form by oxidation of a substrate. The four copper atoms of the cluster are classified into type I, type II and type III. Due to its high redox potential (+790 mV), type I copper is the site responsible for the catalytic oxidation of molecular oxygen into water and plays a significant role in the oxidation of ortho and para diphenols, polyphenols, aminophenols and polyamines.20,21 The schematic representation of the enzyme activity is shown in Scheme 1.
 |
| | Scheme 1 Schematic representation of enzyme activity. | |
There are several reports on the qualitative and quantitative determination of ISP, such as spectrophotometry,22–25 spectrofluorimetry,26–28 high-performance liquid chromatography (HPLC)29,30 and chemiluminescence.31,32 Though they have higher sensitivity and selectivity than electroanalytical methods, they require expensive experimental procedures, and are time consuming and involve complicated solution preparation. Electroanalytical methods such as CV and DPV are simple, rapid and inexpensive analytical techniques used in many fields of chemistry, particularly for the development of new sensors towards the monitoring of drugs, pesticides and environmental pollutants.33–35 In this study we have fabricated an electrochemical laccase biosensor based on multiwalled carbon nanotubes immobilized on a glassy carbon electrode.
To the best of our knowledge, there has been no work reported towards the determination of ISP in serum samples and its simultaneous determination in the presence of UA and AA, using the fabricated Lac-SiSG/MWCNT/GCE. Therefore in this present study, we have demonstrated the electrochemical redox behaviour of ISP at Lac-SiSG/MWCNT/GCE. We have evaluated the quantitative determination of ISP in phosphate buffer solution (PBS), their recoveries in spiked human serum samples and the analytical performance of the developed biosensor towards the simultaneous determination of ISP in the presence of UA and AA.
2. Experimental
2.1 Materials
All materials were received from commercial sources and used without any further purification. Isoprenaline (ISP) and tetraethyl orthosilicate (TEOS) were purchased from Sigma-Aldrich and multiwalled carbon nanotubes (MWCNTs) were from Dropsens, Edificio CEEI, Llanera (Spain). Uric acid (UA), ascorbic acid (AA), K4[Fe(CN)6], Triton X-100 and Na2HPO4 were from Merck specialties Pvt. Ltd, Mumbai and K3[Fe(CN)6], NaH2PO4 were from Fisher Scientific India Pvt. Ltd, Mumbai. The laccase enzyme used in the present study was obtained from applied microbiology laboratory, Department of Virology, S. V. University, Tirupati. This was isolated from fungal culture and from soil contaminated with forest wastes.36
2.2 Instrumentation
Cyclic voltammetry (CV), square wave voltammetry (SWV), chronoamperometry (CA), differential pulse voltammetry (DPV) and electrochemical impedance spectroscopy (EIS) techniques were performed by using a CH–Electrochemical workstation (Model CHI-660D, CH Instruments, Austin, USA) with a conventional three-electrode system comprised of saturated calomel electrode (SCE) as a reference electrode, glassy carbon rod as a counter electrode and Lac-SiSG/MWCNT/GCE as a working electrode. The pH solutions were prepared by mixing 0.1 M NaH2PO4 to 0.1 M Na2HPO4, using an Elico U 120 pH meter combined with a pH CL 51 B electrode for measuring the pH values.
2.3 Preparation of MWCNT and laccase suspension
1 mg of MWCNT was accurately weighed and dissolved in 2 mL of ethanol and sonicated by using an ultrasonication bath for 10 min and stored in a refrigerator when not in use. 10 mg of laccase was accurately weighed and dissolved in 10 mL of phosphate buffer solution at pH 6.5 and used as a laccase enzyme stock solution.
2.4 Preparation of silica sol–gel/laccase enzyme
A homogenous TEOS silica sol–gel was prepared by mixing 4 mL of TEOS, 2 mL of H2O, 100 μL of 0.1 M HCl, 50 μL of 10% Triton-X-100. The mixture was stirred for 1 h to form a clear sol. The sol can be stored for several days when refrigerated. To the mixture of 20 μL of silica sol–gel and 80 μL of PBS pH-6.5, 50 μL of stock laccase enzyme solution was added and the solution was stirred well and stored for further use.37
2.5 Fabrication of Lac-SiSG/MWCNT/GCE
Prier to modification of the glassy carbon electrode (GCE), it was first polished with Al2O3 of 1.0, 0.3 and 0.05 micron size to get a mirror shine. The polished GCE was used as a bare electrode, and then 5 μL of MWCNT suspension was dispersed onto the surface of the bare GCE through physical adsorption and allowed to dry for 5 min to form the MWCNT/GCE.
The obtained MWCNT/GCE was further modified with laccase enzyme by using sol–gel method. 5 μL of Lac-SiSG was immobilized on to the MWCNT/GCE and dried for 1 h. The obtained electrode was washed with buffer solution and it was denoted as Lac-SiSG/MWCNT/GCE.
2.6 Serum sample preparation
The blood sample was collected from a healthy individual aged around 35 years, and was kept aside for 30 min and centrifuged for about 10 min at 3000 rpm. The supernatant serum was collected and stored for further use. To 0.3 mL of serum sample, 3 mL of ethanol was added and centrifuged for 5 min at 3000 rpm, and the obtained protein free serum was collected and used for further analysis.
3. Results and discussion
3.1 Voltammetric characterization of electrodes with K3[Fe(CN)6]
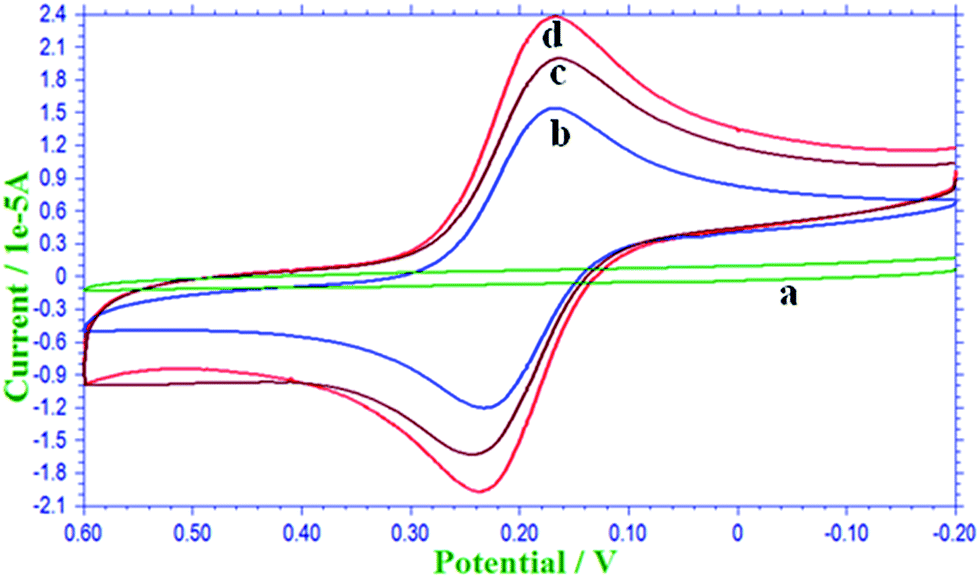
The voltammetric responses of bare GCE, MWCNT/GCE and Lac-SiSG/MWCNT/GCE were examined in 1.0 × 10−3 M [Fe(CN)6]3− in 1 M KCl solution by using the CV technique. Fig. 1 shows the CV response for [Fe(CN)6]3− at bare GCE, MWCNT/GCE and Lac-SiSG/MWCNT/GCE with a scan rate of 0.1 V. At the bare electrode, the peak currents of [Fe(CN)6]3− was iap = −1.452 × 10−5 A with ΔEp = 80 mV. At MWCNT/GCE, the peak current of [Fe(CN)6]3− was enhanced to iap = −1.81 × 10−5 A with decrease in peak separation of ΔEp = 70 mV, revealing a clear indication for the catalytic activity of MWCNT modified GCE. Due to the presence of large surface area and high electrical conductivity properties of MWCNTs, an increase in the peak currents was observed at MWCNT/GCE. Furthermore at Lac-SiSG/MWCNT/GCE, the peak currents of [Fe(CN)6]3− was increased to iap = −2.117 × 10−5 A in comparison with bare GCE and MWCNT/GCE and this was due to the presence of an enhanced catalytic surface area relative to MWCNT/GCE, thus leading to the fast electron transfer. The anodic peak current iap and cathodic peak current icp ratio for [Fe(CN)6]3− at the three different electrodes were nearly unity, indicating the good reversibility of [Fe(CN)6]3−. The effect of scan rate was measured and the peak currents were directly proportional to ν1/2, indicating that the process was under diffusion control. Based on the scan rate results and by employing eqn (1) and (2), we have evaluated the diffusion coefficient (D) and active surface coverage area (Γ) of different electrodes and the values are listed in Table 1.38,39 Here ‘n’ is the number of electrons, ‘C’ is the concentration (mol cm3−), ‘ν’ is the scan rate (V s−1), and ‘F’ is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).| | |
ip = 2.69 × 105n3/2D1/2Cν1/2
| (1) |
 |
| | Fig. 1 CV responses of 1 mM [Fe(CN)6]3− in 0.1 M KCl solution with a scan rate of 100 mV s−1 at bare GCE (‘a’ without [Fe(CN)6]3− and ‘b’ with [Fe(CN)6]3−), MWCNT/GCE (c) and Lac-SiSG/MWCNT/GCE (d). | |
Table 1 Various parameters determined for [Fe(CN)6]3− at three different electrodes
| Electrode |
Diffusion coefficient/cm2 s−1 |
Surface area/mol cm−2 |
iap/icp |
| Do |
Dr |
| Bare GEC |
2.56 × 10−6 |
2.46 × 10−6 |
2.38 × 10−9 |
0.98 |
| MWCNT/GCE |
2.78 × 10−6 |
2.61 × 10−6 |
2.56 × 10−9 |
1.01 |
| Lac-SiSG/MWCNT/GCE |
3.79 × 10−6 |
3.8 × 10−6 |
3.32 × 10−9 |
0.99 |
3.2 EIS characterization of electrodes with ferri/ferro
EIS technique is the most powerful non-destructive and investigative tool for the characterization of the surface nature of different electrodes. The EIS spectrum exhibits semicircular and linear portions; the semicircular part represents the charge transfer resistance (Rct) and linear part describes the low electron transfer rate. In this study we have studied the surface nature of bare GCE, MWCNT/GCE and Lac-SiSG/MWCNT/GCE electrodes at ferri and ferro probe in 1 M KCl solution. Fig. 2a shows the Nyquist plots for bare GCE, MWCNT/GCE and Lac-SiSG/MWCNT/GCE, from which it is evident that the bare electrode showed higher charge transfer resistance (Rct) than the other two electrodes and the Lac-SiSG/MWCNT/GCE showed the least charge transfer resistance, indicating good electron transfer rate at the Lac-SiSG/MWCNT/GCE. Fig. 2b shows the Bode plot for the corresponding bare GCE, MWCNT/GCE and Lac-SiSG/MWCNT/GCE. The equivalent circuit data is listed in Table 2.40
 |
| | Fig. 2 (a) Nyquist plot for bare GCE (a), MWCNT/GCE (b) and Lac-SiSG/MWCNT/GCE (c) in [Fe(CN)6]3−/[Fe(CN)6]4− probe containing in 0.1 M KCl. (b) Bode plot for bare GCE (a), MWCNT/GCE (b) and Lac-SiSG/MWCNT/GCE (c) in [Fe(CN)6]3−/[Fe(CN)6]4− dissolved in 0.1 M KCl. | |
Table 2 EIS data received from circuit fitting for three different electrodes
| Electrode |
Rsol/Ω |
C/μF |
Rct/Ω |
W |
| Bare GCE |
19.09 |
0.86 |
289.4 |
0.00015 |
| MWCNT/GCE |
21.49 |
10.68 |
0.001 |
0.00018 |
| Lac-SiSG/MWCNT/GCE |
21.04 |
16.43 |
0.001 |
0.00014 |
3.3 Electrochemistry of isoprenaline at Lac-SiSG/MWCNT/GCE
To study the electrochemical redox mechanism of ISP, we have recorded CVs of 1 mM ISP in PBS solution of pH 6.5. The scan was performed in three steps between the potential range −0.3 to 0.7 V. The CVs of ISP show that, in the first sweep i.e. from −0.3 V to 0.7 V ISP was found to produce only one oxidation peak at potential ≈0.237 V and this was due to the oxidation of dihydroxy group present in the ISP to the corresponding dione derivative. In the reverse scan i.e. from 0.7 V to −0.3 V, a reduction peak was observed at potential ≈0.202 V which was due to the reversible reduction of dione derivative into ISP. The formed dione derivative at potential ≈0.237 V was found to undergo a 1,4-Michael addition reaction to form a cyclization product that in turn underwent a reduction process at a potential of −0.2 V to form the corresponding dihydroxy cyclization product. In the third (re-oxidation) sweep i.e. from −0.3 to 0.7 V there was an oxidation peak at a potential of −0.16 V which was due to the reversible oxidation of the dihydroxy cyclization product to the dione cyclization product. At the potential 0.162 V a small oxidation peak was observed and this was due to the reductive elimination of a water molecule. The electrochemical redox reaction mechanism is shown in Scheme 2.41–43 Fig. 3 shows the CV responses of ISP at bare GCE, MWCNT/GCE and Lac-SiSG/MWCNT/GCE. From the CV responses, it was observed that there was an increase in the peak currents at MWCNT/GCE in comparison with bare GCE, which was due to the catalytic nature of the MWCNTs immobilized on the surface of the GCE. Further the peak current of ISP at Lac-SiSG/MWCNT/GCE was enhanced in comparison with bare GCE and MWCNT/GCE and this was due to the catalytic nature of copper atoms present in the laccase enzyme.
 |
| | Scheme 2 Electrochemical redox mechanism of isoprenaline. | |
 |
| | Fig. 3 CVs of 1 mM ISP in PBS of pH 6.5 at bare GCE (a), MWCNT/GCE (b) and Lac-SiSG/MWCNT/GCE (c) with a scan rate of 100 mV s−1. | |
3.4 Effect of pH
The effect of pH values of the supporting electrolyte (PBS) ranging from pH 4.5 to 8.0 was studied on the response of peak currents and peak potentials of ISP at Lac-SiSG/MWCNT/GCE with SWV technique. The pH of the supporting electrolyte greatly influenced the peak currents and peak potential response of ISP. It was noticed that when the pH of the electrolyte was increased from lower values, the peak currents of ISP was found to increase up to pH 6.5, above this value the peak currents stared to decline, which may be due to the unavailability of protons in basic medium which leads to unfavorable conditions for the electrochemical redox reaction of ISP. At pH 6.5 we observed highest peak currents of ISP; hence we selected pH 6.5 as optimum pH. The first oxidation peak potentials were found to shift cathodically with increase in the pH of the supporting electrolyte, indicating the involvement of protons in the electrochemical reaction. Fig. 4 shows a graphical representation of peak currents and peak potentials vs. pH and the linear regression equation for the peak potentials against pH was found to be as Ep (V) = 0.527 − 0.061pH. The slope of the linear regression equation was nearly 0.059 V indicating the involvement of equal numbers of protons and electrons.44
 |
| | Fig. 4 Plots of peak currents (■) and peak potentials (●) vs. pH. | |
3.5 Effect of scan rate
In order to determine the kinetic parameters of ISP at Lac-SiSG/MWCNT/GCE, we have studied the effect of scan rates between the ranges from 0.01 V to 0.15 V with CV by taking 1 mM ISP in PBS solution of pH 6.5. It was observed that the peak currents of ISP linearly increased against ν1/2, indicating a diffusion controlled process with linear regression equation of ic1p (μA) = 0.662 (μA) + 17.394ν1/2 (ν in V) with correlation factor r = 0.998. Fig. 5a shows CVs of ISP with different scan rates from 0.01 to 0.15 V and Fig. 5b is the linear plot of peak current against ν1/2.45,46 Based on eqn (3) and (4) we have determined the charge transfer coefficient ‘α’ as 0.39 and heterogeneous rate constant ‘ks’ as 1.41 × 103− cm−1. Here ‘n’ is the number of electrons involved in the rate determining step and ‘α’ is the charge transfer coefficient, ‘D’ is the diffusion coefficient and ‘m’ is the slope of Ep vs. lnν.| | |
Ep = E0 − m[0.78 + ln(D1/2/ks) + (m/2)(lnm)] + m/2lnν
| (3) |
 |
| | Fig. 5 (a) CV scans of 1 mM ISP in PBS solution of pH 6.5 at Lac-SiSG/MWCNT/GCE with scan rates of 10 mV s−1 (a), 20 mV s−1 (b), 30 mV s−1 (c), 40 mV s−1 (d), 50 mV s−1 (e), 60 mV s−1 (f), 70 mV s−1 (g), 80 mV s−1 (h), 90 mV s−1 (i), 100 mV s−1 (j), 110 mV s−1 (k), 120 mV s−1 (l), 130 mV s−1 (m), 140 mV s−1 (n) and 150 mV s−1 (o). (b) Plot of the current (μA) vs. square root of scan rate, ν1/2 (V s−1)1/2). | |
3.6 Effect of concentration
CA is the more sensitive technique which gives information about the peak currents at low concentration levels. In this study we have studied the concentration effect on the peak currents of ISP in PBS at Lac-SiSG/MWCNT/GCE. The slope values of CA curves were found to linearly increase with increase in the concentration of the ISP. Fig. 6a shows the chronoamperometric responses of different ISP concentrations and the inset is a magnified view. Fig. 6b shows the linear dependence of slope values from CA against the concentration of ISP ranging from 5 × 10−6 M to 6 × 10−4 M and the linear regression equations were found to be as follows
| Slope = 5.65 + 0.064C (μM) |
| Slope = 6.16 + 0.086C (μM) |
 |
| | Fig. 6 (a) Chronoamperometric responses of ISP with different concentrations: 4 μM (a), 7 μM (b), 11 μM (c), 15 μM (d), 25 μM (e), 35 μM (f), 50 μM (g), 70 μM (h), 100 μM (i), 140 μM (j), 200 μM (k), 250 μM (l), 300 μM (m), 350 μM (n), 400 μM (o), 500 μM (p) and 600 μM (q) at Lac-SiSG/MWCNT/GCE. (b) Plot between the slope values of chronoamperometric curves and their peak currents. | |
The limit of detection (LOD) and limit of quantification (LOQ) values were evaluated from the slopes of above equations and using eqn (5) and (6), where ‘S’ is the standard deviation and ‘m’ is the slope for the calibration curve, the LOD and LOQ values were 1.8 × 10−7 M and 6.0 × 10−7 M, respectively.47 The LOD and linear detection range (LDR) values of ISP with other methods are compared in Table 3.
Table 3 Comparison table for the determination of ISP with different methods
| Method |
Detection |
LDR/μM |
LOD/μM |
Ref. |
| Cyclic voltammetry. High performance liquid chromatography. Differential pulse voltammetry. Copper(II) hexacyanoferrate(III). |
| CVa |
CuHCFd |
196–1070 |
80 |
2 |
| Spectrophotometry |
Silica reactor |
123–738 |
62.5 |
26 |
| Chemiluminescence |
— |
0.94–236 |
0.236 |
31 |
| HPLCb |
Chemiluminescence |
— |
0.00016 |
48 |
| DPVc |
1-Butyl-3-methylimidazolium hexafluorophosphate |
1.0–520.0 |
0.85 |
49 |
| DPV |
Lac-SiSG/MWCNT/GCE |
50.0–250.0 |
0.18 |
Present work |
3.7 Simultaneous determination of ISP in the presence of UA and AA
The simultaneous determination of ISP in the presence of biologically important molecules such as UA and AA is a challenging task, because the oxidation potentials of ISP, UA and AA are very close to each other. The selectivity and resolution of ISP, UA and AA at the Lac-SiSG/MWCNT/GCE is of great interest. Therefore the main objective of the present study was to determine ISP in the presence of UA and AA in PBS buffer and as well as PBS buffer spiked with serum samples. The fabricated electrode can resolve well all the three compounds separately with good sensitivity in comparison with MWCNT/GCE.
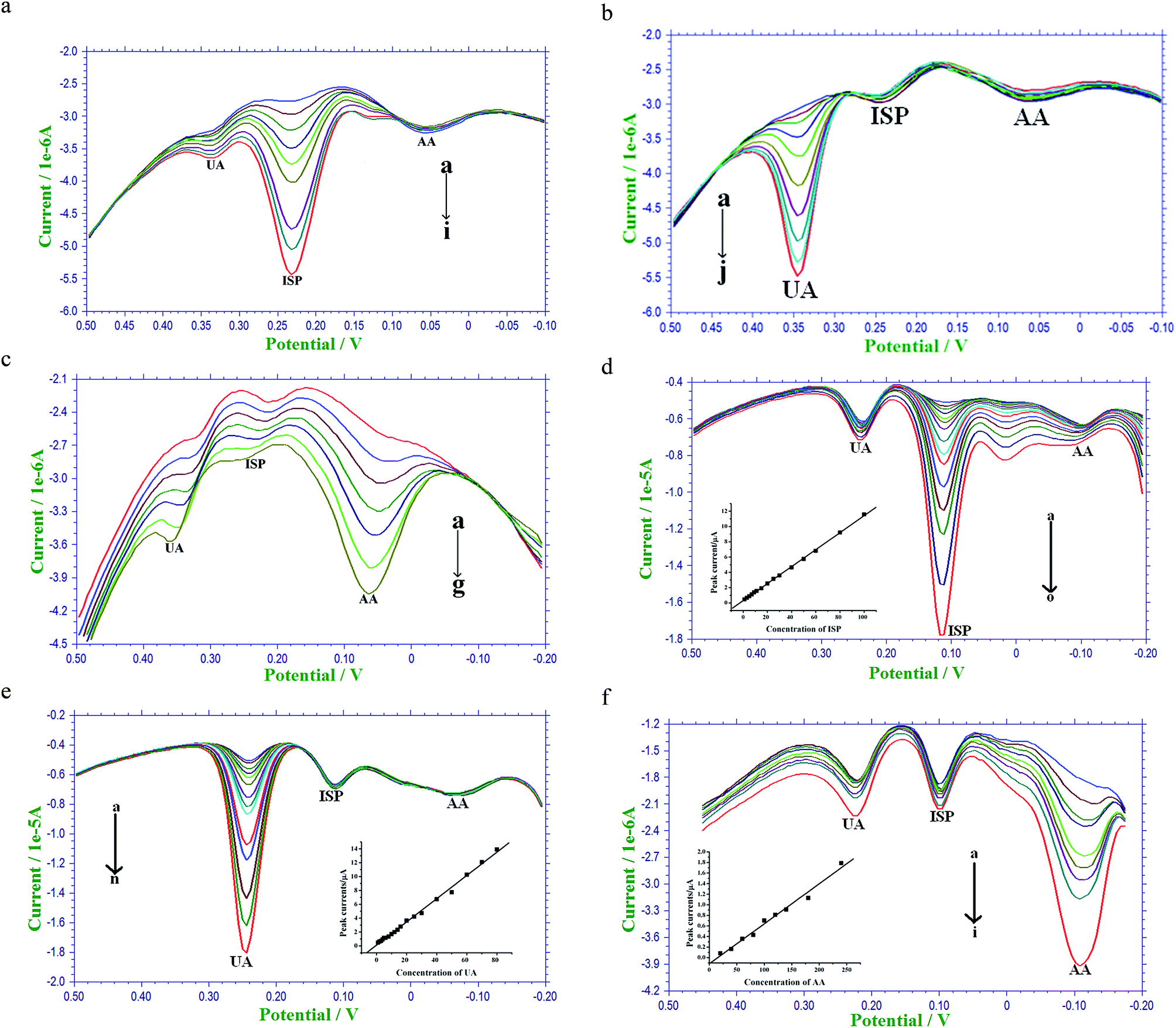
Fig. 7a shows the increase in the peak currents of ISP in PBS buffer at potential ≈0.2 V with a constant increase in the ISP concentration from 5 μM to 45 μM, in the presence of UA (4 μM) and AA (200 μM), from the figure we could see that there was no influence of ISP concentration on the UA and AA responses. Fig. 7b shows the constant increase of peak currents of UA with increase in the concentration from 5 μM to 50 μM in the presence of constant concentrations of ISP (12 μM) and AA (200 μM), from this result we note that there was no influence of UA concentration on the ISP and AA. Fig. 7c shows the increase in the peak currents of AA with the constant increase in the concentration from 50 μM to 350 μM in the presence of constant concentration of ISP (4 μM) and UA (4 μM), from this figure we note that there was a slight positive shift in the peak potentials of ISP and UA, this may be due to the more acidic nature of AA. As the concentration of AA was increased, the pH of the solution changes to lower pH values and causes the shift. We have studied the non-interfering concentration range of UA and AA against ISP, where we note that there was no interference of UA concentration from 20–1000 μM against 10 μM of ISP and also on further increase of UA concentration beyond 1000 μM there was no influence on the electrochemical signal tendency of ISP, on the other hand as the concentration of AA was increased there was an influence on the ISP signal.
 |
| | Fig. 7 (a) DPVs corresponding to simultaneous determination of ISP with different concentrations: 5 μM (a), 10 μM (b), 15 μM (c), 20 μM (d), 25 μM (e), 30 μM (f), 35 μM (g), 40 μM (h), 45 μM in the presence of UA (4 μM) and AA (200 μM) at Lac-SiSG/MWCNT/GCE. (b) DPVs corresponding to simultaneous determination of UA with different concentrations: 5 μM (a), 10 μM (b), 15 μM (c), 20 μM (d), 25 μM (e), 30 μM (f), 35 μM (g), 40 μM (h), 45 μM (i) and 50 μM (j) in the presence of ISP (12 μM) and AA (200 μM) at Lac-SiSG/MWCNT/GCE. (c) DPVs corresponding to simultaneous determination of AA with different concentrations: 50 μM (a), 100 μM (b), 150 μM (c), 200 μM (d), 250 μM (e), 300 μM (f), 350 μM (g) in the presence of UA (4 μM) and ISP (4 μM) at Lac-SiSG/MWCNT/GCE. (d) DPVs corresponding to ISP in PBS buffer spiked with human serum samples with different concentrations: 1 μM (a), 3 μM (b), 5 μM (c), 7 μM (d), 9 μM (e), 11 μM (f), 15 μM (g), 20 μM (h), 25 μM (i), 30 μM (j), 40 μM (k), 50 μM (l), 60 μM (m), 80 μM (n) and 100 μM (o), in the presence of UA (10 μM) and AA (150 μM) at Lac-SiSG/MWCNT/GCE. (e) DPVs corresponding to UA in PBS buffer spiked with human serum samples with different concentrations: 6 μM (a), 8 μM (b), 10 μM (c), 12 μM (d), 14 μM (e), 16 μM (f), 20 μM (g), 25 μM (h), 30 μM (i), 40 μM (j), 50 μM (k), 60 μM (l), 70 μM (m), 80 μM (n) in the presence of ISP (20 μM) and AA (150 μM) at Lac-SiSG/MWCNT/GCE. (f) DPVs corresponding to AA in PBS buffer spiked with human serum samples with different concentrations: 20 μM (a), 40 μM (b), 60 μM (c), 80 μM (d), 100 μM (e), 120 μM (f), 140 μM (g), 180 μM (h) and 240 μM (i) in the presence of UA (4 μM) and ISP (4 μM) at Lac-SiSG/MWCNT/GCE. | |
Fig. 7d shows the increase in the peak currents at potential ≈0.1 V for ISP in PBS buffer spiked with human serum sample. ISP concentration was increased gradually from 1 μM to 100 μM, in the presence of UA (10 μM) and AA (150 μM), a linear relation was observed between the peak currents and different concentrations of ISP, the linear equation was found to be as ip (μA) = 0.3056 (μA) + 0.0111CISP (μM). Based on the linear equation the limit of detection value was determined as 2.3 μM. Fig. 7e shows the increase of peak currents of UA with increase in the concentrations of UA ranging from 6 μM to 80 μM under constant concentrations of ISP (20 μM) and AA (150 μM) and the inset of the figure is the plot between the peak currents and different concentrations of UA, a linear equation was observed as ip (μA) = 0.1108 (μA) + 0.0168CUA (μM) and the detection limit for UA in the presence of ISP and AA was 4.7 μM. Fig. 7f shows the increase in the peak currents of AA with increase in the concentration from 20 μM to 240 μM with constant concentration of ISP (5 μM) and UA (10 μM) and the inset of the figure is the plot drawn between the peak currents and concentrations of AA, a linear equation ip (μA) = −0.1163 (μA) + 0.0076CAA (μM) was observed and from the linear equation the limit of detection value was 27.4 μM. From Fig. 7c, we could clearly see that there was an influence of AA on the ISP and UA in the pure PBS buffer, but whereas from Fig. 7f, we could see that there was no much influence of AA on the ISP and UA in PBS buffer spiked with human serum samples, this may be due to the change in the pH of the supporting electrolyte.
3.8 Recoveries from pharmaceutical formulation and serum samples
The developed method was effectively used for the determination of ISP in injection samples and the concentration of ISP in the injection samples was verified by using standard addition method. Firstly, the collected ISP injection sample was diluted to required concentration and the same concentration was prepared with the standard drug sample. The recoveries for different concentrations of injection samples were in the range from 101% to 104% and the values are listed in Table 4. The recoveries of ISP injection in serum samples were studied by using the standard addition method and are also listed in Table 4. The recovery values suggested that the proposed biosensor was satisfactory.
Table 4 Determination of ISP in ISP injection samples at serum and buffer solutions
| Medium |
Added/μM |
Found/μM |
Recovery (%) |
Bias (%) |
| Buffer |
40 |
41.7 |
104.25 |
+4.25 |
| 60 |
61.1 |
101.83 |
+1.83 |
| 80 |
80.6 |
100.75 |
+0.75 |
| |
| Serum sample |
40 |
39.31 |
98.28 |
−1.72 |
| 60 |
57.2 |
95.33 |
−4.67 |
| 80 |
77.5 |
96.88 |
−3.12 |
3.9 Repeatability, reproducibility and stability
To investigate the practical utilization of the fabricated biosensor, we have studied repeatability, reproducibility and stability. The developed biosensor was tested with 1 mL of 10 mM ISP in 9 mL of pH 6.5 PBS using CV and the responses were recorded several times. The CV responses of each repeated cycle for ISP was nearly the same and had a low RSD value, i.e., 4.22% indicating good reproducibility of the fabricated biosensor. Fig. 8 shows the CVs of 1 mM ISP in PBS pH-6.5 with the scan rate of 100 mV s−1 and the inset was the plot between the peak currents and number of measurements.
 |
| | Fig. 8 CVs of 30 replications corresponding to 1 mM ISP in PBS solution of pH 6.5 with a scan rate of 100 mV s−1. Insert is the graphical representation of measurement number against peak current. | |
The biosensor electrode was separately prepared four times and the CV responses recorded for 1 mL of 10 mM ISP in 9 mL of pH-6.5 PBS. The current responses of the four different electrodes were almost the same showing small relative standard deviation (RSD).
The proposed biosensor was continuously tested for 50 successive cycles with a scan rate of 100 mV s−1 in solution containing 1 mL of 10 mM ISP in 9 mL of pH 6.5 PBS. The relative standard deviation of the CV responses suggested that the developed biosensor was found to have good stability.46
4. Conclusions
Herein, we have demonstrated the electrochemical redox behaviour of ISP at a Lac-SiSG/MWCNT/GCE biosensor. The fabricated biosensor was found to exhibit a good electrochemical catalytic ability to determine ISP with low detection limits. The fabricated biosensor had the capability for the determination of ISP in the presence of UA and AA simultaneously. Moreover the practical applicability of the developed electrode was examined in human serum samples with satisfactory results. Finally this study will facilitate a simple and versatile protocol for the monitoring of ISP concentrations with good repeatability, reproducibility and stability.
Acknowledgements
One of the authors P. Gopal gratefully acknowledges the University grant commission (UGC) for providing financial support through Basic scientific research (BSR)-Research fellowship for meritorious students.
References
- H. Beitollahi and I. Sheikhshoaie, Electrochim. Acta, 2011, 56, 10259–10263 CrossRef CAS PubMed.
- V. G. Bonifacio, L. H. Marcolino Jr, M. F. S. Teixeira and O. F. Filho, Microchem. J., 2004, 78, 55–59 CrossRef CAS PubMed.
- M. Chen, X. Ma and X. Li, J. Solid State Electrochem., 2012, 16, 3261–3266 CrossRef CAS.
- A. Kutluay and M. Aslanoglu, Acta Chim. Slov., 2010, 57, 157–162 CAS.
- C. Bian, Q. Zeng, H. Xiong, X. Zhang and S. Wang, Bioelectrochemistry, 2010, 79, 1–5 CrossRef CAS PubMed.
- Y. Ni, M. Wei and S. Kokot, Int. J. Biol. Macromol., 2011, 49, 622–628 CrossRef CAS PubMed.
- A. A. Ensafi and H. K. Maleh, Int. J. Electrochem. Sci., 2010, 5, 1484–1495 CAS.
- L. S. Goodman and A. Gilman, The Pharmacological Basis of Therapeutics, McGraw-Hill, New York, 9th edn, 1996, pp. 105–120 Search PubMed.
- N. Pearce and M. J. Hensley, Epidemiol. Rev., 1998, 20, 173–186 CrossRef CAS.
- Code of Federal Regulations, Title 21, Food and Drugs: Parts 200–299.
- D. Han, T. Han, C. Shan, A. Ivaska and L. Niua, Electroanalysis, 2010, 22, 2001–2008 CrossRef CAS.
- J. Ping, J. Wu, Y. Wang and Y. Ying, Biosens. Bioelectron., 2012, 34, 70–76 CrossRef CAS PubMed.
- M. M. Ardakani, L. Hosseinzadeh, A. Khoshroo, H. Naeimi and M. Moradian, Electroanalysis, 2014, 26, 275–284 CrossRef.
- H. Beitollahia and I. Sheikhshoaieb, Electrochim. Acta, 2011, 56, 10259–10263 CrossRef PubMed.
- A. A. Ensafi, M. Dadkhah and H. K. Maleh, Colloids Surf., B, 2011, 84, 148–154 CrossRef CAS PubMed.
- A. A. Ensafi, H. Bhrami, H. K. Maleh and S. Mallakpour, Chin. J. Catal., 2012, 33, 1919–1926 CrossRef CAS.
- X. Yang, B. Feng, X. He, F. Li, Y. Ding and J. Fei, Microchim. Acta, 2013, 180, 935–956 CrossRef CAS.
- M. Keyvanfard, V. Khosravi, H. K. Maleh, K. Alizad and B. Rezaei, J. Mol. Liq., 2013, 177, 182–189 CrossRef CAS PubMed.
- B. J. Sanghavi and A. K. Srivastava, Electrochim. Acta, 2011, 56, 4188–4196 CrossRef CAS PubMed.
- D. Monti, G. Ottolina, G. Carrea and S. Riva, Chem. Rev., 2011, 111, 4111–4140 CrossRef CAS PubMed.
- C. Tortolini, S. Rea, E. Carota, S. Cannistraro and F. Mazzei, Microchem. J., 2012, 100, 8–13 CrossRef CAS PubMed.
- H. Jeon, H. Nohta, H. Nagaoka and Y. Ohkura, Anal. Sci., 1991, 7, 257 CrossRef CAS.
- H. Nohta, T. Yukizawa, Y. Ohkura, M. Yoshimura, J. Ishida and M. Yamaguchi, Anal. Chim. Acta, 1997, 344, 233 CrossRef CAS.
- G. Alberts, T. Lameris, A. H. V. Meiracker, A. J. M. I. Veld and F. Boomsma, J. Chromatogr., Biomed. Appl., 1999, 730, 213 CrossRef CAS.
- R. M. V. Camanas, J. M. S. Mallols, J. R. T. Lapasio and V. R. Ramos, Analyst, 1995, 120, 1767 RSC.
- K. O. Lupetti, I. C. Vieira and O. F. Filho, Talanta, 2002, 57, 135 CrossRef CAS.
- P. Solich, C. K. Polydorou, M. A. Koupparis and C. E. Efstathiou, J. Pharm. Biomed. Anal., 2000, 22, 781 CrossRef CAS.
- J. J. B. Nevado, J. M. L. Gallego and P. B. Laguna, Anal. Chim. Acta, 1995, 300, 293 CrossRef.
- Y. Qu, L. F. Moons and F. Vandesande, J. Chromatogr., Biomed. Appl., 1997, 704, 351 CrossRef CAS.
- F. Mashige, Y. Matsushima, C. Miyata, R. Yamada, H. Kanazawa, I. Sakuma, N. Takai, N. Shinozuka, A. Ohkubo and K. Nakahara, Biomed. Chromatogr., 1995, 9, 221 CrossRef CAS PubMed.
- G. J. Zhou, G. F. Zhang and H. Y. Chem, Anal. Chim. Acta, 2002, 463, 257 CrossRef CAS.
- C. Zhang, J. Huang, Z. Zhang and M. Aizawa, Anal. Chim. Acta, 1998, 374, 105 CrossRef CAS.
- P. V. Narayana, T. M. Reddy, P. Gopal, K. Reddaiah and P. Raghu, Res. J. Chem. Sci., 2014, 4, 37–43 CAS.
- P. Raghu, T. M. Reddy, K. Reddaiah, B. E. K. Swamy and M. Sreedhar, Food Chem., 2014, 142, 188–196 CrossRef CAS PubMed.
- P. Raghu, M. R. M. Reddy, T. M. Reddy, B. E. K. Swamy and K. Reddaiah, Anal. Bioanal. Electrochem., 2012, 4, 1–16 Search PubMed.
- B. Viswanath, B. Rajesh, A. Janardhan, A. Praveen Kumar and G. Narasimha, Enzyme Res., 2014, 2014 DOI:10.1155/2014/163242.
- P. Raghu, B. E. K. Swamy, T. M. Reddy, B. N. Chandrashekar and K. Reddaiah, Bioelectrochemistry, 2012, 83, 19–24 CrossRef CAS PubMed.
- K. Reddaiah, T. M. Reddy, P. Raghu and P. Gopal, Anal. Bioanal. Electrochem., 2012, 4, 372–385 Search PubMed.
- P. Raghu, T. M. Reddy, K. Reddaiah, L. R. Jaidev and G. Narasimha, Enzyme Microb. Technol., 2013, 52, 377–385 CrossRef CAS PubMed.
- K. Reddaiah, M. M. Reddy, P. Raghu and T. M. Reddy, Colloids Surf., B, 2013, 106, 145–150 CrossRef CAS PubMed.
- R. N. Goyal, A. R. S. Rana and H. Chasta, Electrochim. Acta, 2012, 59, 492–498 CrossRef CAS PubMed.
- Z. Wang, Z. Zhang, Z. Fu, L. Fang, W. Luo, D. Chen and X. Zhang, Anal. Chim. Acta, 2003, 494, 63–70 CrossRef CAS.
- K. Reddaiah, T. M. Reddy and P. Raghu, J. Electroanal. Chem., 2012, 682, 164–171 CrossRef CAS PubMed.
- K. Reddaiah, T. M. Reddy, Y. S. Rao, P. Raghu and P. Gopal, Mater. Sci. Eng., B, 2014, 183, 69–77 CrossRef CAS PubMed.
- P. Gopal, T. M. Reddy, K. Reddaiah, P. Raghu and P. V. Narayana, J. Mol. Liq., 2013, 178, 168–174 CrossRef CAS PubMed.
- P. Raghu, T. M. Reddy, P. Gopal, K. Reddaiah and N. Y. Sreedhar, Enzyme Microb. Technol., 2014, 57, 8–15 CrossRef CAS PubMed.
- J. Uhrovcik, Talanta, 2014, 119, 178–180 CrossRef CAS PubMed.
- M. Yamaguchi, J. Ishida and M. Yoshimura, Analyst, 1998, 123, 307 RSC.
- A. A. Ensafi and H. Karimi-Maleh, Drug Test. Anal., 2011, 3, 325–330 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.