
Mesoporous graphitic carbon nitride as a heterogeneous catalyst for photoinduced copper(I)-catalyzed azide–alkyne cycloaddition†
Sajjad Dadashi-Silaba,
Baris Kiskana,
Markus Antoniettib and
Yusuf Yagci*ac
aDepartment of Chemistry, Istanbul Technical University, 34469 Maslak, Istanbul, Turkey. E-mail: yusuf@itu.edu.tr; Fax: +90 212 285 6386; Tel: +90 212 285 3241
bDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Potsdam, D-14424, Germany
cCenter of Excellence for Advanced Materials Research (CEAMR) and Department of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, 21589, Saudi Arabia
First published on 10th October 2014
Abstract
A new protocol for the photoinduced copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) was developed. The system utilizes mesoporous graphitic carbon nitride (mpg-N3C4) as a heterogeneous photocatalyst to realize the in situ formation of the required copper(I) catalyst by reducing copper(II) species upon UV or sunlight irradiation. The efficiency of the system in regard to its ability to photoinitiate the CuAAC of a variety of aliphatic and aromatic azides and alkynes in different solvents was examined. Photocalorimetric measurements confirmed that the rate of the reaction depends on the light intensity and continuous or intermittent irradiation. Importantly, because of the heterogeneous nature of the photocatalyst, mpg-C3N4 can easily be separated and reused in further reactions without loss of activity.
The concept of “Click Chemistry” introduced by the groups of Sharpless1 and Meldal2 has opened new possibilities in the field of synthetic chemistry. Since then, many studies have been devoted towards better understanding the philosophy of “Click Chemistry” and expanding its scope to various fields of chemistry ranging from bioconjugation3–7 and drug discovery8 to material science9–12 and many others.13
Copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) has been a very prime example of click chemistry that is catalyzed and influenced by copper(I) salts (Cu(I)) towards regioselectivity. Supplementing various promoters or ligands within the system enhances the catalytic performance of Cu(I) compounds and accelerates the rate of reaction. Typical examples include benzimidazole and phosphoramidite ligands, acids and bases and polymeric ligands as reusable catalyst.14–19
The required Cu(I) to catalyze CuAAC can also be formed in an in situ manner by reducing the air stable Cu(II) species using reducing agents,20 electrochemical,21,22 or photochemical23 protocols. In the photochemical path, the process is accomplished either by direct reduction of Cu(II)/ligand complexes by light24–26 or indirect reduction by the photochemically generated active species mostly free radicals.27,28 Bowman and co-workers employed the indirect reduction system to achieve a comprehensive spatiotemporal control over the CuAAC process.29 Multi-functional azide and alkyne groups patterned areas in contact with light where the photogenerated radicals reduced Cu(II) to Cu(I). Undoubtedly, the ability to provide spatiotemporal control over the reaction is a unique advantage of the photoinduced click reactions over the other conventional processes. We have recently shown that the spectral sensitivity of such systems can be extended to higher wavelengths by using polynuclear aromatic compounds (PAC).30 The electron transfer reaction of photoexcited PAC with Cu(II) within the exciplex generates Cu(I) species that trigger the CuAAC process. An alternative approach was reported independently by the groups of Bowman and Vincent who incorporated acylphosphinate oxide and benzophenone-like photoinitiators, respectively, within the Cu(II) catalyst as the counter anion. In the case of acylphosphinate, the CuAAC click reaction was catalyzed by the photochemically generated electron donor radicals stemming from acylphosphinate.31 In the case of benzophenone photosensitizer, the catalytic action involving Cu(I) generation is also based on the photoinduced electron transfer process. It was proposed that quenching the triplet aromatic ketone, most likely by the ligand, led to the formation of ligand centered radical cation followed by hydrogen abstraction to yield ultimately the required Cu(I) complex.32,33 Quite recently, Bear et al. reported an elegant method for doping copper(I) cations within ZnS quantum dots (QDs) shells.34 The CuAAC was achieved by the release of Cu(I) ions into solution upon photoexcitation of the QDs. Interestingly, the system operated in a ligand-free mode, and the heterogeneous QDs being refined the Cu dopant exhibited reusability for further usages in catalyzing the CuAAC. Readers are directed towards more detailed and specific click reactions triggered by light.35
The polymeric, metal-free, semiconducting photocatalyst mesoporous graphitic carbon nitride (mpg-C3N4),36 known as a “green” catalyst, is meanwhile used in a broad variety of photocatalytic applications due to its large surface area (≈200 m2 g−1), surface active sites, high stability, and easily separation from the system. The reported photocatalyst applications include hydrogen evolution by water splitting,37 reduction of CO2,38,39 Friedel–Crafts reaction,40 oxidation of benzene,41 and photoinitiation of free radical polymerization.42 We have recently shown that atom transfer radical polymerization (ATRP) can successfully be realized by photochemical means using mpg-C3N4 for catalyst activation.43 The proposed ATRP mechanism is based on the in situ generation of Cu(I) species by the reduction of Cu(II) with the photochemically released electrons from the semiconductor mpg-C3N4. To further explore and take advantage of this type of redox process, it was the intention here to develop a methodology for CuAAC, which is similarly activated by Cu(I) ions to potentially arrive at a green and reusable click process.
In this communication, we report that a highly efficient CuAAC process can be realized by the photochemical formation of Cu(I) ions in a controlled manner without the need of a reducing agent or radical initiator by using the metal free heterogeneous catalyst mpg-C3N4.
We began with a model reaction between benzyl azide (1a) and phenylacetylene (2a) in the presence of copper(II) chloride/N,N,N′,N′,N′′-pentamethyldiethylenetriamine (CuIICl2/PMDETA) and mpg-C3N4 under UV light irradiation (Fig. 1-a). The reaction was monitored by FTIR as the azide peak decreased throughout the clicking of 1a and 2a. The reaction completed after 45 min irradiation as the azide peak around 2100 cm−1 completely disappeared (Fig. 1-b). The copper catalyst was used at a ratio of 0.05 equiv. The effect of the ratio of Cu(II) was further investigated by loading it at the ratios of 0.01, 0.05, and 0.10 equiv. The kinetics of the reactions, which were monitored by FTIR, showed that by using Cu(II) at the ratio of 0.01, the reaction reached only to 81% yield after 90 min irradiation. Whereas in others increasing the ratio of the Cu increased the rate of the reaction reaching to 99% of conversion within 45 min for the ratio of 0.05 and 30 min for 0.10 equiv. (Fig. 1-c). Although utilizing Cu at the ratio of 0.10 gave high conversion in a low reaction time, because of element economy it was chosen to proceed with the ratio of 0.05, which gave the same conversion in relatively low irradiation time. Control experiments in the absence of light or mpg-C3N4 gave only traces of the final triazole product in the same irradiation time as that of utilizing mpg-C3N4. In addition, the model reaction was also examined under natural sunlight irradiation and it proceeded to completion within 80 min.
 | ||
| Fig. 1 Kinetics of the photoinduced CuAAC using mpg-C3N4: (a) schematic representation of photoinduced clicking benzyl azide (1a) with phenylacetylene (2a) by mpg-C3N4; (b) typical presentation of decreasing the intensity of the azide peak in 1a as a result of clicking with 2a monitored by FTIR (0.05 equiv. of Cu(II)); (c) yield versus irradiation time for clicking 1a with 2a calculated from FTIR spectra loading different copper ratios. | ||
A series of experiments were carried out to further explore the effect of light intensity on the process. For this purpose, we turned to photodifferential scanning calorimetry (photo-DSC) to investigate the calorimetric behavior of the photoinduced CuAAC between the model substitutes 1a and 2a (see ESI†). The obtained thermograms provide a real-time measure of the reaction rate (Fig. 2). As anticipated, the rate of the reaction increased by increasing the light intensity. Bowman et al. found that the intensity of light had no significant influence on the rate of click reaction between multi-functional azide and alkyne groups to afford polymers containing triazole rings. It was attributed to the fact that the effect of light was confined to the very first step of the reaction that is to photolysis of the initiator to form free radicals capable of reducing Cu(II).44 However, in our system it is obvious that the intensity of light has a noticeable influence on the rate and yield of the reaction as presented in Fig. 2-a. Apparently, more intense light facilitates the formation of surface localized charge carriers to reduce Cu(II).
 | ||
| Fig. 2 Calorimetric traces of the photoinduced CuAAC in the presence of mpg-C3N4 deploying benzyl azide (1a) and phenylacetylene (2a) as the model substitutes: (a) effect of light intensity on the rate of the photoinduced CuAAC (inset: real time conversion of the reaction), and (b) effect of continuous and intermittent irradiation on the rate of the reaction measured by photo-DSC. | ||
Fig. 2-b shows the calorimetric measurements of the system in which two samples were exposed to light; one under continuous irradiation and the other for only 3 min. The samples were kept in the dark for initially 2 min and no heat change was observed indicating the necessity of light in the initiation step. Exposing to light started the reaction where by turning the light off (after 3 min irradiation) the rate of the reaction dropped dramatically yet continued to occur in a lower rate with respect to the continuous irradiation. This persistence of the reaction in the dark implies that the in situ formed Cu(I) species are not completely consumed by oxidation/reduction processes. An integration under the heat–time graph revealed that the conversion of the sample under continuous irradiation was 1.2 times greater than that of the 3 min irradiated sample.
The scope of the reaction was examined for a variety of azide groups using 2a as the alkyne part (Scheme 1). Tolerance for variations in the azide component is excellent. As shown in Scheme 1, the system worked out well for all tested azides having different functionalities. When an azide with highly aromatic group, anthracene (1b), with respect to phenyl ring (1a), was incorporated, the reaction rate increased significantly. Although the reaction with phenyl azide (1c) proceeded rather slowly, acceleration was observed when fluoro (1d) or nitro (1e) groups were attached at the para position. This was in line with the previously reported studies that revealed electron withdrawing or donating substituents on the azide substrate result in a rate increase.45 In addition, ester functionalized (1f) and unsaturated azides (1g) reacted fully with 2a.
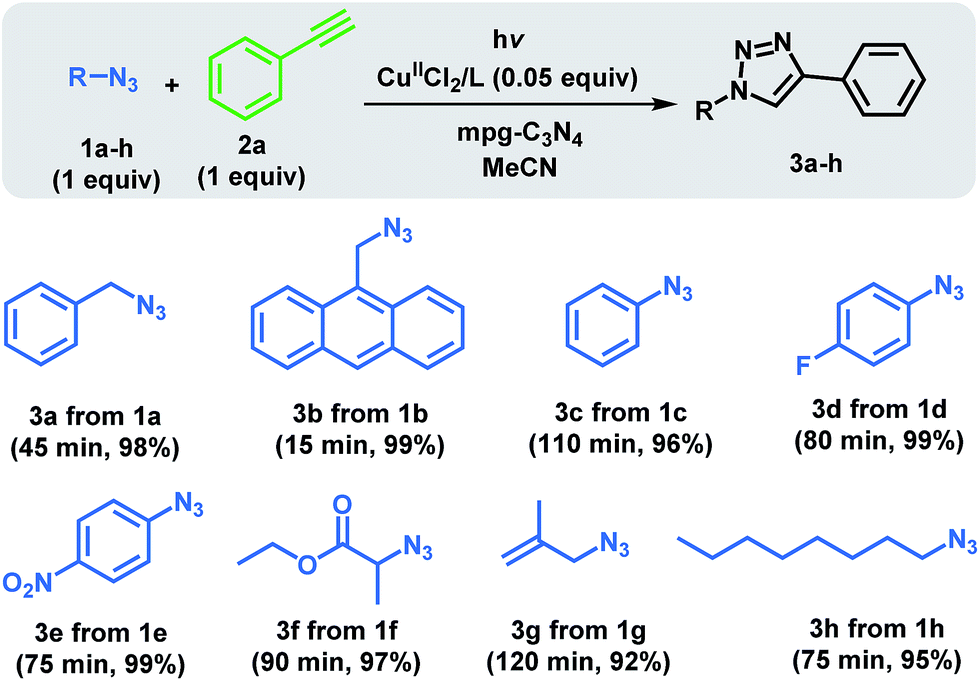 | ||
| Scheme 1 Azide scope for the photoinduced CuAAC using mpg-C3N4. | ||
The applicability of the system was also investigated for various alkyne compounds applying 1a as the azide component (Scheme 2). Compared with 2a, the rate of clicking slightly decreased in the case of pyridine (2b) and thiophene (2c) substitutes, but still reacted in relatively short time with high yields. In the case of propargyl moieties, propargylamine (2e) and propargyl alcohol (2f) reacted rather slowly. Notably, the reaction with propargyl acrylate (2d) was fast with high yield.
 | ||
| Scheme 2 Alkyne scope for the photoinduced CuAAC using mpg-C3N4. | ||
The model reaction proceeded quite well in a variety of solvents, especially in MeCN in accordance with the previous studies.46 However, the reaction was rather inefficient in alcohols and water (Table S1†). Importantly, the heterogeneous nature of this system affords easily filtration of the photocatalyst off the system and utilizing it for further reactions. The model reaction was repeated five times loading the same mpg-C3N4 photocatalyst. The yield of the reaction remained almost constant for all cycles indicating preservation of the photocatalyst during the photoinduced CuAAC (Fig. S2†). FTIR analysis further proved this structural as the spectral behavior of mpg-C3N4 before and after irradiation was the same (Fig. S3†).
Conclusions
In conclusion, we have demonstrated the CuAAC process stimulated by the photoinduced electrons to the in situ formation of Cu(I) to catalyze the click reaction. The photoreduction process was realized by using mpg-C3N4 under UV light in which the heterogeneous nature of the photocatalyst made it to be successfully used for several times for the initiation of CuAAC. It was found by photocalorimetric measurements that the rate of the process is highly reliant on the intensity of light. The system showed an efficacious performance for a wide range of azides and alkynes in several media.Acknowledgements
The authors thank Istanbul Technical University Research Fund for financial support.Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, J. Am. Chem. Soc., 2003, 125, 3192 CrossRef CAS PubMed.
- R. Breinbauer and M. Kohn, ChemBioChem, 2003, 4, 1147 CrossRef CAS PubMed.
- J. F. Lutz and H. G. Borner, Prog. Polym. Sci., 2008, 33, 1 CrossRef CAS PubMed.
- D. Odaci, B. N. Gacal, B. Gacal, S. Timur and Y. Yagci, Biomacromolecules, 2009, 10, 2928 CrossRef CAS PubMed.
- S. Doran, E. Murtezi, F. B. Barlas, S. Timur and Y. Yagci, Macromolecules, 2014, 47, 3608 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS PubMed.
- W. X. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052 CrossRef CAS.
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 176 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC.
- C. Shao, X. Wang, Q. Zhang, S. Luo, J. Zhao and Y. Hu, J. Org. Chem., 2011, 76, 6832 CrossRef CAS PubMed.
- L. S. Campbell-Verduyn, L. Mirfeizi, R. A. Dierckx, P. H. Elsinga and B. L. Feringa, Chem. Commun., 2009, 16, 2139 RSC.
- V. O. Rodionov, S. I. Presolski, S. Gardinier, Y. H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696 CrossRef CAS PubMed.
- C. Shao, X. Wang, J. Xu, J. Zhao, Q. Zhang and Y. Hu, J. Org. Chem., 2010, 75, 7002 CrossRef CAS PubMed.
- L. Bonami, W. Van Camp, D. Van Rijckegem and F. E. Du Prez, Macromol. Rapid Commun., 2009, 30, 34 CrossRef CAS PubMed.
- M. Lammens, J. Skey, S. Wallyn, R. K. O'Reilly and F. Du Prez, Chem. Commun., 2010, 46, 8719 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- N. K. Devaraj, P. H. Dinolfo, C. E. D. Chidsey and J. P. Collman, J. Am. Chem. Soc., 2006, 128, 1794 CrossRef CAS PubMed.
- V. Hong, A. K. Udit, R. A. Evans and M. G. Finn, ChemBioChem, 2008, 9, 1481 CrossRef CAS PubMed.
- S. C. Ritter and B. Koenig, Chem. Commun., 2006, 4694 RSC.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945 CrossRef CAS PubMed.
- B. Sandmann, B. Happ, J. Vitz, M. D. Hager, P. Burtscher, N. Moszner and U. S. Schubert, Polym. Chem., 2013, 4, 3938 RSC.
- A. A. Alzahrani, A. H. Erbse and C. N. Bowman, Polym. Chem., 2014, 5, 1874 RSC.
- M. A. Tasdelen, G. Yilmaz, B. Iskin and Y. Yagci, Macromolecules, 2012, 45, 56 CrossRef CAS.
- Y. Yagci, M. A. Tasdelen and S. Jockusch, Polymer, 2014, 55, 3468 CrossRef CAS PubMed.
- B. J. Adzima, Y. H. Tao, C. J. Kloxin, C. A. DeForest, K. S. Anseth and C. N. Bowman, Nat. Chem., 2011, 3, 256 CrossRef CAS PubMed.
- G. Yilmaz, B. Iskin and Y. Yagci, Macromol. Chem. Phys., 2014, 215, 662 CrossRef CAS.
- T. Gong, B. J. Adzima and C. N. Bowman, Chem. Commun., 2013, 49, 7950 RSC.
- L. Harmand, S. Cadet, B. Kauffmann, L. Scarpantonio, P. Batat, G. Jonusauskas, N. D. McClenaghan, D. Lastecoueres and J.-M. Vincent, Angew. Chem., Int. Ed., 2012, 51, 7137 CrossRef CAS PubMed.
- L. Harmand, R. Lambert, L. Scarpantonio, N. D. McClenaghan, D. Lastecoueres and J.-M. Vincent, Chem.–Eur. J., 2013, 19, 16231 CrossRef CAS PubMed.
- J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930 CrossRef CAS PubMed.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- M. B. Ansari, B. H. Min, Y. H. Mo and S. E. Park, Green Chem., 2011, 13, 1416 RSC.
- F. Goettmann, A. Thomas and M. Antonietti, Angew. Chem., Int. Ed., 2007, 46, 2717 CrossRef CAS PubMed.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2006, 45, 4467 CrossRef CAS PubMed.
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658 CrossRef CAS PubMed.
- B. Kiskan, J. S. Zhang, X. C. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546 CrossRef CAS.
- S. Dadashi-Silab, M. A. Tasdelen, B. Kiskan, X. C. Wang, M. Antonietti and Y. Yagci, Macromol. Chem. Phys., 2014, 215, 675 CrossRef CAS.
- T. Gong, B. J. Adzima, N. H. Baker and C. N. Bowman, Adv. Mater., 2013, 25, 2024 CrossRef CAS PubMed.
- V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705 CrossRef CAS PubMed.
- B. R. Buckley, S. E. Dann, D. P. Harris, H. Heaney and E. C. Stubbs, Chem. Commun., 2010, 46, 2274 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, additional spectral data and supplementary figures and table. See DOI: 10.1039/c4ra09954k |
| This journal is © The Royal Society of Chemistry 2014 |