
Rational design and synthesis of novel 2-(substituted-2H-chromen-3-yl)-5-aryl-1H-imidazole derivatives as an anti-angiogenesis and anti-cancer agent†
Abstract
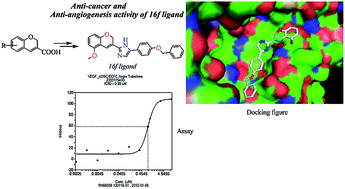
Based on earlier proven pharmacophore analogues of cancer a novel 2-(substituted-2H-chromen-3-yl)-5-aryl-1H-imidazoles (13–16) were rationally designed and synthesized by the reaction of chromene-3-carboxylic acids (10a–d) with substituted acyl bromides in the presence of TEA followed by refluxing with NH4OAc in toluene. Compounds 13–16 were screened in vitro for the inhibition of KRAS/Wnt and their anti-angiogenesis properties. Compound 16f has been identified as a potent anti-angiogenesis molecule, which can be considered as a new lead structure. The molecular docking analysis displayed the higher binding affinity of 16f with KRAS, Wnt and VEGF.
 Please wait while we load your content...
Please wait while we load your content...

