
Carborane polyimides, synthesis and characterization†
Tao Xing*,
Yuhong Huang,
Kai Zhang and
Juying Wu
Institute of System and Engineering, Chinese Academy of Engineering Physics, Mianyang 621900, China. E-mail: 412xingt@caep.ac.cn; Fax: +86 816 2486494; Tel: +86 816 2486494
First published on 8th October 2014
Abstract
Here we report the syntheses of polyimides with meta-carborane cages embedded in the polymer chain and their characterizations. Aromatic carborane diamines were prepared by a Sonogashira reaction in the first place; reaction of the obtained diamines with commercially available dianhydride led to the formation of carborane polyimides with high molecular weight. Thermogravimetric analysis (TGA) of the samples showed that they have excellent thermal stability, with 5 wt% decomposition temperatures (Td5%) higher than 520 °C. Due to the boron-rich nature of the obtained polyimides, samples prepared here have potential applications in the aerospace industry and nuclear power stations as building blocks for neutron shielding and detecting devices.
Introduction
Preparation of functional polymeric materials with superior physical and chemical properties is an ever existing goal for scientists and engineers. Polyimide material, since its discovery in 1908, has been a focus of scientific research ever since. Now industrialized polyimide products such as polyimide film and resin are commercially available and widely used in the microelectronic and aerospace industries.1,2 Efforts have been made both to address old problems of this highly conjugated and less soluble polymer3–5 and to develop new functional polyimide materials with potential future applications.6–9 With decades of efforts, various functional polyimides have now been developed to meet various applications.Neutrons are neutral particles which are usually employed in places like nuclear power station and hospital for purposes like energy production and tumour therapy.10,11 Uncharged nature of neutron makes this kind of radiation particularly difficult to shield. Neutron usually travels through object and reacts with nucleus of atoms. Exposure to neutrons usually leads to severe tissue damage due to a denser ion path as neutrons lose/deposit their energy within the target material and the production of secondary radiations such as γ and α particles.12 Thus the development of effective shielding gears with superior physical properties is particularly important for workers in nuclear power station and aerospace. Neutron shielding materials currently developed are mainly composites prepared by blending particles such as boron carbide with matrix polymeric materials such as epoxy resin,13 polypropylene and polyethylene.14,15 Application of these materials are restricted due to the inferior physical property such as mechanical and thermal property of the matrix resin, while under some conditions such as in aerospace shielding materials with higher thermal property are desired. Besides, due to the absence of chemical interaction between matrix and boron particles, boron particles blended in the polymer matrix can easily be removed by washing. Thus the development of shielding polymeric materials with more effective shielding property and superior physical property is vital for their application in specific fields such as aerospace.
Icosahedral carboranes (C2B10H12) are boron rich clusters with exceptional thermal and chemical stability.16–18 High boron content of these clusters opens door for the preparation of materials with neutron-trapping ability.19,20 The rationale of this lies in the fact that neutron undergoes nuclear reaction with 10B to produce α particles, 7Li and γ ray (eqn 1).
| 10B + 1n → 4He(α) + 7Li + 2.4 MeV | (1) |
This unique property makes these compounds useful in areas such as boron neutron therapy and neutron shielding.13,21,22 Preparation of carborane polymer thus provides a way for the preparation of polymer with neutron shielding ability. Of all the carborane polymers reported, work describing synthesis and characterization of carborane polyimide is rather scarce, our search about this new area only yielded one paper describing the synthesis of carborane contained poly(ester-imide)s.23 They were prepared by condensation of phenylhydroquinone bistrimethylsilyl ether phenylhydroquinone and dicarboxylic acid chloride at high temperature, and no data relevant with the thermal stability is provided in this article.
Here in this manuscript we report carborane polyimides prepared by a traditional diamine/dianhydride condensation method and their characterization. Aromatic carborane diamines were prepared by Sonogashira reaction in the first place, reaction of the obtained diamines with commercially available dianhydride led to the formation of carborane polyimides with high molecular weight. All the carborane polyimides obtained could form flexible films by a solution casting method. Thermogravimetric analysis of the samples shows that they have high thermal stability, with Td5%s higher than 520 °C. Due to the introduction of high boron content carborane cage into the polyimide backbone, carborane polyimides obtained here have potential application in aerospace and nuclear power station as building blocks for neutron shielding and detecting devices.
Results and discussion
Aromatic carborane diamine syntheses
Sonogashira reaction catalyzed by palladium(0) between aryl and terminal alkynes, is a widely employed method for the preparation of arylalkyne.24 Since its compatibility with functional groups such as hydroxyl and amine group,25,26 this method could generate aromatic diamines while at the same time eliminate those usually used reduction reaction related with aromatic amine preparation. Aromatic carborane diamines used here were both prepared by a one-step Sonogashira reaction between aromatic dihalide and para-ethynylaniline (Scheme 1). Aromatic dihalides 1 and 3 used here could readily be prepared by Cu-catalyzed coupling reaction or nucleophilic substitution reaction. Et3N was used both as co-solvent and base for the Sonogashira reaction, aromatic carborane diamine in both cases were obtained in moderate yields after column chromatography. Compared with 1, compound 3 with iodobenzene moiety shows relatively higher reactivity, the coupling reaction thus could smoothly progress at room temperature. Structures of these two new aromatic diamines were confirmed by FT-IR, 1H NMR, 13C NMR and mass spectra (Fig. S1–S8†).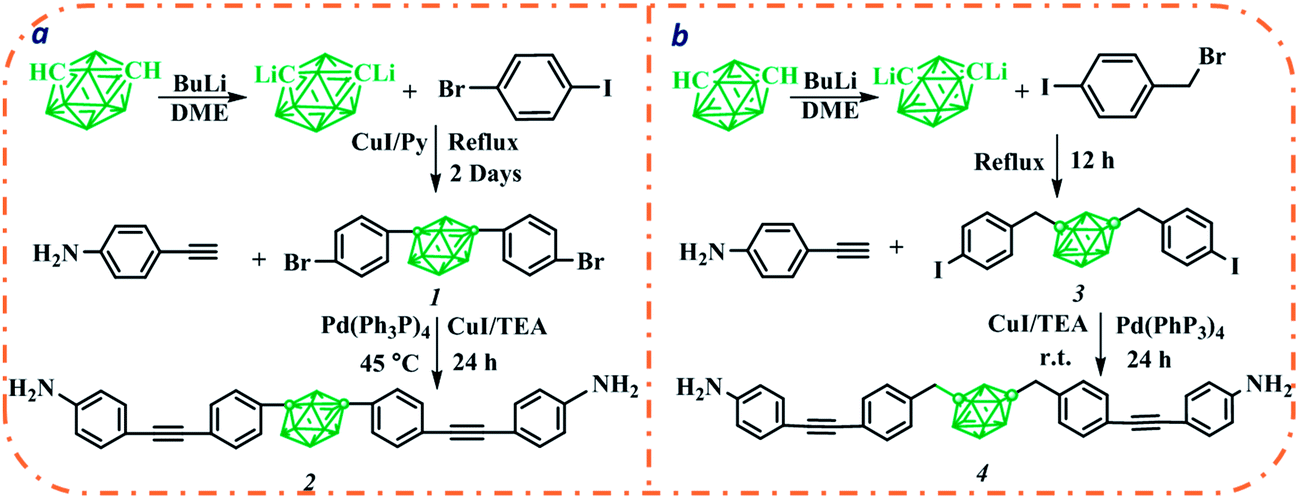 | ||
| Scheme 1 Synthesis of aromatic carborane diamines. | ||
Carborane polyimide syntheses
These new carborane polyimides were all synthesized by a two-step process. 4,4′-diaminodiphenyl ether was also copolymerized to yield carborane polyimides with different carborane content (Scheme 2). The recipe was shown in Table 1 (Column 2). Reaction between aromatic carborane diamines and commercially available 6FDA led to the formation of carborane polyamic acids which were later imidized by a chemical imidization method using acetic anhydride/pyridine. For the preparation of polyamic acids, all reactions proceeded rapidly and generated viscous polyamic acid solutions within 3 h. Inherent viscosity measurements of the resulted polyamic acid solutions indicated the formation of polymer with high molecular weight (Table 1). All reactions were stopped at 15 h and purified by precipitating into methanol. Molecular weights and polydispersity indexes of the obtained carborane polyimides were determined by GPC measurements and the results were shown in Table 1. All carborane polyimides obtained have molecular weights higher than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, indicating that the carborane diamines prepared here have high reactivity. The polyimide solution casted on a glass plate could readily form flexible film after evaporation of solvent by heating.
000 g mol−1, indicating that the carborane diamines prepared here have high reactivity. The polyimide solution casted on a glass plate could readily form flexible film after evaporation of solvent by heating.
 | ||
| Scheme 2 Synthesis of carborane polyimides. | ||
| Entry | Recipe | Tg/°C | Td5%/°C | ηinh (30 °C)/dL g−1 | Mw/104 | Mw/Mn | d/μm | |
|---|---|---|---|---|---|---|---|---|
| In air | In N2 | |||||||
| a Mole ratio, diamine 2:ODA:6FDA for b, d and f; diamine 4:ODA:6FDA for a, c and e.b Thickness of the film prepared by solution casting method. |
||||||||
| a | 1:0:1a |
206 | 572 | 536 | 0.59 | 2.3 | 1.2 | 4.2b |
| b | 1:0:1 |
227 | 549 | 539 | 1.29 | 7.5 | 1.1 | 4.6 |
| c | 1:1:2 |
220 | 545 | 539 | 0.66 | 2.4 | 1.7 | 5.0 |
| d | 1:1:2 |
226 | 549 | 541 | 0.63 | 3.3 | 2.3 | 5.8 |
| e | 1:2:3 |
224 | 570 | 537 | 1.05 | 6.0 | 1.3 | 3.6 |
| f | 1:2:3 |
221 | 533 | 529 | 0.63 | 3.4 | 1.8 | 3.4 |
Chemical structure of the obtained polyimide films were studied by FI-IR measurements. The results were shown in Fig. 1.
 | ||
| Fig. 1 FT-IR spectra of the obtained polyimide films. | ||
All the films show intensive absorptions in the infrared spectra at 2581–2615 cm−1 which are characteristic stretching vibrations of the BH-groups in the closo-polyhedron.27 Bands at 1780 (asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) and 1731 cm−1 (symmetric CO stretching) correspond to the imide rings along polyimide main chain.28 Adsorption bands at 2200 cm−1 are characteristic bands of alkyne, indicating the alkyne groups embedded in the polyimide chain are stable under polyimide synthesis and the subsequent drying process.
O stretching) and 1731 cm−1 (symmetric CO stretching) correspond to the imide rings along polyimide main chain.28 Adsorption bands at 2200 cm−1 are characteristic bands of alkyne, indicating the alkyne groups embedded in the polyimide chain are stable under polyimide synthesis and the subsequent drying process.
Chemical compositions of the obtained polyimides were all determined by elemental analysis. The results were shown in Table 2. As can be seen in Table 2, theoretical element content is close to the measured value in all cases, indicating that the diamine monomers are of high purity and the polymerization of the dianhydride and diamine was complete in each case, polyimides of the expected chemical composition were obtained after reaction.
Physical property
UV-Vis transmittance spectra of the films were shown in Fig. 2. The thickness of the film used for measurement was shown in Table 1. For both series of carborane polyimides with carborane diamine 2 and 4 as starting materials, decrease the feed ratio of carborane diamine resulted in films with lower transparency, carborane polyimides with the highest amount of ODA show the lowest transparency. This is probably because the bulky carborane is helpful for the disruption of charge transfer between polyimide chains, which is assumed to be responsible for the usually deep colour of polyimide film.2 One another reason is that the carborane cage is strongly electron-withdrawing,29 introducing carborane effectively weakens electron donating ability of the diamines, thus suppresses the intermolecular charge-transfer complexing (CTC) between alternating electron donor (diamine residue) and electron acceptor (dianhydride residue) moieties. | ||
| Fig. 2 UV-Vis transmittance spectra of the polyimide films. | ||
Thermal properties of the obtained polyimides were evaluated by both differential scanning calorimetry (DSC) and TG, the results are shown in Table 1. Glass transition temperatures of the obtained polyimides are all around 220 °C, relatively lower Tgs of these new polyimides could be attributed to the carborane cages embedded in the main chain. The bulky cage reduces chain-to-chain charge transfer interactions and results in less dense chain packing. Thus the segments of the chain could move at relatively lower temperature.30 DSC curves also show endothermal peaks above 350 °C (Fig. 3). This is due to the cross-linking reaction of alkyne groups embedded in the main chain. The peak shifts to higher temperature with the decreasement of carborane diamine. This is because the introduction of ODA dilutes the alkyne density on the polymer chain, the cross-linking reaction could only happen at higher temperature under dilute conditions.31 DSC curves also show exothermal peaks around 550 °C, which are resulted from the degradation of the polymer at these temperatures.
 | ||
| Fig. 3 DSC curves of the obtained carborane polyimides in N2. | ||
Polyimide samples dried at 50 °C under vacuum show premature weight loss caused by the evaporation of solvent trapped in the sample during chemical imidization (Fig. 4). No premature weight loss was observed after drying the sample at 200 °C under vacuum. TG analysis of the obtained polyimides in air showed similar thermo stability as they did in N2, Td5%s of all samples are above 520 °C (Table 1), weight losses of these samples starting at 500 °C are caused by the simultaneous decomposition of m-carborane and imide groups, this is proved by the disappearance of both bands corresponding to imide ring (1780 and 1731 cm−1) and BH groups (2580 cm−1) after heating the polyimides to 500 °C in dry air and keeping at this temperature for 3 h (Fig. S9†). This may also explain why all samples show similar thermal stability irrespective of their carborane content.
 | ||
| Fig. 4 TGA curves of the obtained carborane polyimide in N2. | ||
Wide-angle X-ray diffraction (WAXRD) patterns of the obtained polyimide films b, d and f were shown in Fig. 5, from which we can see that all films obtained were non-crystalline materials. Non-crystalline nature of these films could be attributed to the carborane moiety introduced into the polyimide main chain, which disrupts intermolecular packing. The originally broad peak becomes sharper as carborane content decreased (from b to f), indicating a non-crystalline to crystalline transition as carborane content was reduced.
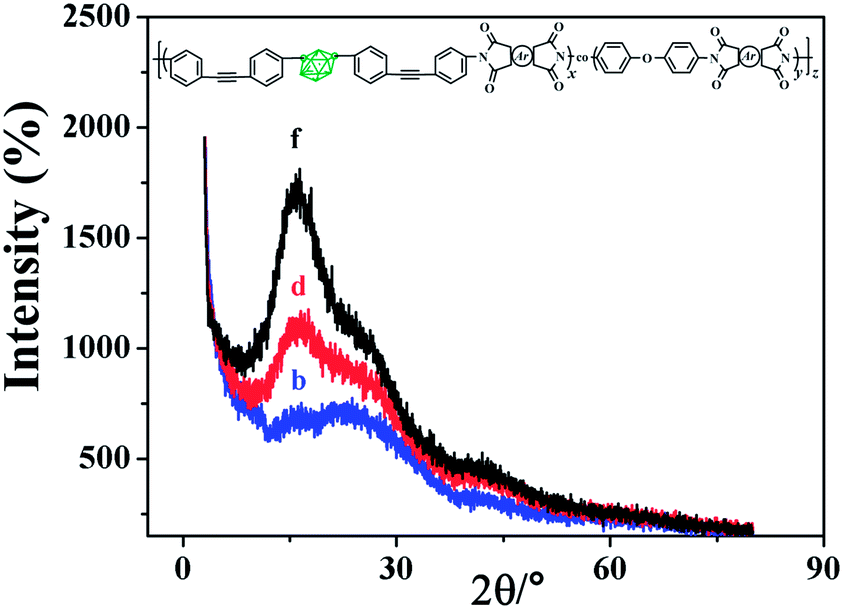 | ||
| Fig. 5 WAXRD curves of the carborane polyimide films. | ||
Experimental
Materials
Compounds n-BuLi (1.6 M in hexane), Pd(PPh3)4, cuprous iodide (CuI), meta-carborane, triethylamine (TEA), 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA) and di-(4-aminophenyl)ether (ODA) were purchased from J&K scientific Ltd. Para-ethynylaniline was purchased from TCI Corporation. Pyridine (Py), dimethoxyethane (DME) and N,N′-dimethylformamide (DMF) were purchased from Aladdin corporation and dried with calcium hydride before use. Silica gel (200–400 mesh) was purchased from Qindao Haiyang Chemical Institute. Palladium solution (0.1% PdCl2 in 3 M HCl (aq.)) was used for spot developing of thin layer chromatography (TLC), which gives black spot upon heating. All other reagents were purchased from Aladdin Corporation and used without purification.Characterization
1H and 13C NMR were recorded on a Bruker Avance 300 or 400 MHz NMR spectrometer as indicated. Both deuterated chloroform (CDCl3) and dimethyl sulfoxide (DMSO) were used as solvent. 1H NMR spectra were referenced to residual protio impurity in the solvent (CDCl3, 7.26 ppm, DMSO, 2.51 ppm). 13C NMR spectra were referenced to the solvent resonance (CDCl3 77.0 ppm, DMSO, 39.6 ppm). Chemical shifts were reported in ppm, and the coupling constants were reported in Hz. IR spectra were recorded in the range 400–4000 cm−1 on a Perkin Elmer frontier spectrometer using a KBr pellet method. For the liquid sample, a drop of the sample was dipped onto KBr pellet before measurement. Thermogravimetric (TG) and differential scanning calorimetry (DSC) analyses were performed on a Perkin Elmer STA-8000 DSC-TGA analyzer. All thermal experiments (TGA and DSC) were carried out with heating rates of 10 °C min−1 and a nitrogen or air flow rate of 20 mL min−1. UV-Vis spectra were obtained on a Shimadzu UV-2401PC Ultraviolet spectrophotometer. Molecular weights of the samples were determined by Gel Permeation Chromatography (GPC) equipped with two columns (one Shodex GPC KD-804 column and one guard column), a refractive index detector (RID-10A), DMF was used as the mobile phase and the measurement was performed at 60 °C at a sample concentration of 2 mg mL−1. Monodispersed polystyrene standards were used for the calibration of Mn, Mw and Mw/Mn. polyamic acid solutions (0.5 g dL−1 in DMF) were used for viscosity measurement. The measurements were performed at 30 °C with an Ubbelohde viscometer. The wide-angle X-ray diffraction (WAXRD) measurements were undertaken on a Philos X-ray diffractometer with Cu–K radiation (40 kV, 30 mA) from 5–90° with a scanning rate of 2° min−1. Element analysis of the sample was performed on a Elementar Vario EL cube elemental analyzer. Mass spectra were recorded on a Finnigan TSQ Quantum Ultra mass spectrometer.Conclusions
Aromatic carborane diamines were synthesized in the first place by one-step Sonogashira reaction. The obtained carborane diamines were polymerized with 6FDA to prepare carborane polyimides, ODA was also used as an ingredient to generate carborane polyimides with different structure. The high molecular weight polyimides obtained are amorphous materials and of high thermal stability. Due to a combination of high thermal stability and boron-riched structure, carborane polyimides prepared here have potential application in future neutron shielding fields such as aerospace and nuclear power station.Acknowledgements
The National Science Foundation of China (no. 201404095) is greatly acknowledged for its financial support. Chinese Academy of Engineering Physics is also appreciated for financial support (no. 2014B0302024).Notes and references
- M. Ding, Prog. Polym. Sci., 2007, 32, 623 CrossRef CAS.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907 CrossRef CAS.
- D. Sahadeva Reddy, C.-H. Chou, C.-F. Shu and G.-H. Lee, Polymer, 2003, 44, 557 CrossRef CAS.
- C.-H. Chou, D. S. Reddy and C.-F. Shu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3615 CrossRef CAS.
- F. Han, M. Ding and L. Gao, Polymer, 1999, 40, 3809 CrossRef CAS.
- K. Morita, K. Ebara, Y. Shibasaki, M. Ueda, K. Goto and S. Tamai, Polymer, 2003, 44, 6235 CrossRef CAS.
- X. Jin and H. Ishii, J. Appl. Polym. Sci., 2006, 100, 4240 CrossRef CAS.
- Y. Shoji, R. Ishige, T. Higashihara, S. Kawauchi, J. Watanabe and M. Ueda, Macromolecules, 2010, 43, 8950 CrossRef CAS.
- T. Suzuki, Y. Yamada and Y. Tsujita, Polymer, 2004, 45, 7167 CrossRef CAS.
- E. Calzada, F. Grünauer, B. Schillinger and H. Türck, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 651, 77 CrossRef CAS.
- Y. Huang, W. Zhang, L. Liang, J. Xu and Z. Chen, Chem. Eng. J., 2013, 220, 143 CrossRef CAS.
- S. Nambiar and J. T. W. Yeow, ACS Appl. Mater. Interfaces, 2012, 4, 5717 CAS.
- N. Hayashi, N. Ishihara and Y. Tasaka, Neutron shielding material composition, shielding material and container, U.S. Patents 7803288 B2, 2010.
- C. Harrison, E. Burgett, N. Hertel and E. Grulkel, Polyethylene/Boron Containing Composites for Radiation Shielding Applications, in Nanostructured Materials and Nanotechnology II: Ceramic Engineering and Science Proceedings, Volume 29, Issue 8, John Wiley & Sons, Inc, 2009, p. 77 Search PubMed.
- C. Harrison, S. Weaver, C. Bertelsen, E. Burgett, N. Hertel and E. Grulke, J. Appl. Polym. Sci., 2008, 109, 2529 CrossRef CAS.
- C. Wang, Y. Zhou, F. Huang and L. Du, React. Funct. Polym., 2011, 71, 899 CrossRef CAS.
- A. González-Campo, B. Boury, F. Teixidor and R. Núñez, Chem. Mater., 2006, 18, 4344 CrossRef.
- M. A. Fox and K. Wade, J. Mater. Chem., 2002, 12, 1301 RSC.
- X. Q. Pan, H. Wang, S. Shukla, M. Sekido, D. M. Adams, W. Tjarks, R. F. Barth and R. J. Lee, Bioconjugate Chem., 2002, 13, 435 CrossRef CAS PubMed.
- H. Koganei, M. Ueno, S. Tachikawa, L. Tasaki, H. S. Ban, M. Suzuki, K. Shiraishi, K. Kawano, M. Yokoyama, Y. Maitani, K. Ono and H. Nakamura, Bioconjugate Chem., 2012, 24, 124 CrossRef PubMed.
- J. Lichtenhan, X. Fu and S. LeClair. Polyhedral oligomeric silsesquioxanes and metallized polyhedral oligomeric silsesquioxanes as coatings, composites and additives, U.S. Patent 20050192364, 2005.
- X. Fu, J. D. Lichtenhan and P. Wheeler, Neutron shielding composition, U.S. Patent 20090085011, 2008.
- H. R. Krichhdori, C. Bruhn, A. Russanov and L. Komarova, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 279 CrossRef.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- G. Vives, J. M. Guerrero, J. Godoy, S. Khatua, Y.-P. Wang, J. L. Kiappes, S. Link and J. M. Tour, J. Org. Chem., 2010, 75, 663 CrossRef PubMed.
- O. S. d. R. Barros, A. Favero, C. W. Nogueira, P. H. Menezes and G. Zeni, Tetrahedron Lett., 2006, 47, 2179 CrossRef CAS.
- X. Zhang, L. Kong, L. Dai, X. Zhang, Q. Wang, Y. Tan and Z. Zhang, Polymer, 2011, 52, 4777 CrossRef CAS.
- D.-J. Liaw, B.-Y. Liaw, L.-J. Li, B. Sillion, R. Mercier, R. Thiria and H. Sekiguchi, Chem. Mater., 1998, 10, 734 CrossRef CAS.
- P. Ducrot and C. Thal, Tetrahedron Lett., 1999, 40, 9037 CrossRef CAS.
- S.-H. Hsiao, H.-M. Wang, W.-J. Chen, T.-M. Lee and C.-M. Leu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3109 CrossRef CAS.
- M. K. Kolel-Veetil, H. W. Beckham and T. M. Keller, Chem. Mater., 2004, 16, 3162 CrossRef CAS.
- R. Coult, M. A. Fox, W. R. Gill, P. L. Herbertson, J. A. H. MacBride and K. Wade, J. Organomet. Chem., 1993, 462, 19 CrossRef CAS.
- R. Djeda, J. Ruiz, D. Astruc, R. Satapathy, B. P. Dash and N. S. Hosmane, Inorg. Chem., 2010, 49, 10702 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, FT-IR and mass spectra of compound 2 and 4, FT-IR spectra of polyimide b, d and f after heating to 500 °C in air for 3 h. See DOI: 10.1039/c4ra09886b |
| This journal is © The Royal Society of Chemistry 2014 |