
Features of the potential energy surface for the SiO + OH → SiO2 + H reaction: relationship to oxygen isotopic partitioning during gas phase SiO2 formation
Abstract
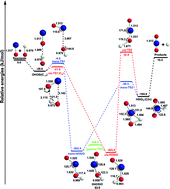
The SiOOH potential energy surface has become central to the understanding of recent experiments (Science 2013, 342, 463) by Chakraborty associated with nebular meteorite formation. The entrance complex, transition states, and exit complex for the title reaction SiO + OH→ SiO2 + H have been studied using the CCSD(T) method with correlation consistent basis sets as large as cc-pV(Q+d)Z. Reported here are characteristics of the reactants, products, six transition states, and four intermediate complexes for this reaction. These show four previously undiscovered stationary point geometries. The entrance complex OH⋯OSi is predicted to lie 28.6 kJ mol−1 below the separated reactants. The classical barriers cis-TS1 and trans-TS1 are predicted to lie 21.8 kJ mol−1 and 6.8 kJ mol−1, respectively, below the reactants. The exit complex HSiO2 is bound by 115.3 kJ mol−1 relative to the separated products. After zero-point vibrational energy corrections, the reaction energy is predicted to be −1.4 kJ mol−1. Vibrational frequencies of the stationary points are reported and compared with the limited available experimental results. The SiOOH potential surface is found to be very different from that for COOH, contrary to the analogy drawn by Chakraborty. Notwithstanding, the assumption of Chakraborty appears justified, because all the stationary points for the SiO + OH reaction have lower relative energies than known for the analogous carbon system.
 Please wait while we load your content...
Please wait while we load your content...

