
Polymer micelle-based combination therapy of paclitaxel and resveratrol with enhanced and selective antitumor activity
Mengying Hub,
Jinfang Zhubd and
Liyan Qiu*ac
aMinistry of Education (MOE) Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, 38 Zheda Road, Hangzhou 310027, China. E-mail: lyqiu@zju.edu.cn; Fax: +86-571-87952306; Tel: +86-571-87952306
bCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China
cCollaborative Innovation Center for Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China
dCollege of Food Science and Pharmaceutical Science, Xinjiang Agricultural University, Urumqi 830052, China
First published on 17th November 2014
Abstract
Owing to the low therapeutic efficacy and high toxicity associated with conventional single drug chemotherapy, combination therapy has been adopted in clinics. We employed polymer micelles for sequential delivery of resveratrol (RES) and paclitaxel (PTX) to obtain a synergistic anticancer activity. Briefly, PTX and RES co-encapsulated micelles were prepared by a thin film method using mPEG-b-PLA block copolymers as carriers with a particle diameter of 20 nm and high encapsulation efficiencies of 95% for both PTX and RES. Furthermore, time-dependent sequential release of two drugs resulted in sensitizing the cancer cells to apoptosis. In vitro cytotoxicity tests the combination strategy exerted a synergistic effect (combination index, CI = 0.7–0.8) both in the PTX-resistant human lung adenocarcinoma epithelial (A549/T) cell line and mice sarcoma 180 (S180) cells while showing very limited toxicity towards the normal human hepatic (L02) cell strain and normal human kidney (HK-2) cell line, which could be illustrated from manipulating ROS levels by redox modulation and reducing protective autophagy. In vivo, the combination therapy achieved the best antitumor effect in all treatment groups in S180 bearing mice and evoked the most significant changes in the cytoarchitecture leading to tumor regression according to the histological studies. In conclusion, PTX and RES co-encapsulated micelles would provide a potential strategy to selectively treat cancers by inducing ROS-dependent apoptosis and inhibiting autophagy.
1. Introduction
Cancer has been one of the greatest threats to public health around the world involving various genetic alterations and cellular abnormalities, which leads to significant mortality in patients.1 Although chemotherapy is still one of the main options for cancer treatments besides surgery and radiation therapy, conventional single drug chemotherapy is commonly associated with low therapeutic efficacy and high toxicity due to limited accessibility of the drug to tumor tissues2 as well as the development of multi-drug resistance (MDR) after repeated treatment.3 Therefore, combination therapy has been adopted in clinics. Generally, combination therapy refers to two or more therapeutic agents delivered simultaneously or a combination of different therapies,4 among which co-delivery of different chemotherapeutic agents is the most common strategy to overcome side-effects resulted from high doses of single drugs and sensitize tumor cells to chemotherapeutic agents as a consequence of synergistic action.Inspiredly, we focused on the combination therapy of seriously toxic paclitaxel (PTX) and resveratrol (RES) which has been reported to possess broad-spectrum beneficial health effects.5
PTX is one of the most widely-used chemotherapeutic agents, and has been demonstrated significant antitumor activity in clinic against a broad range of solid tumors, especially against non-small-cell lung cancer, metastatic breast cancer and refractory ovarian cancer.6 For the mechanism, it is generally believed that PTX is the first generation of microtubule stabilizing agent, which disrupts the formation of normal spindles at the metaphase of cell division, resulting in apoptotic cell death. However, there are common severe side effects including kidney damage7 during PTX treatment along with the development of drug-resistance.
Herein, we tried to introduce RES into the formulations as an effective chemosensitizers8,9 to enhance anti-tumor efficacy and reduce adverse side effects of PTX. RES is a natural polyphenolic abundant in food sources such as grapes, peanuts and red wine.10 Previous studies have shown that RES has a strong chemopreventive effect against the development of cancers of skin, breast, prostate and lung11,12 via a ROS-dependent apoptosis pathway13 accompanied with reduced anti-apoptotic BCl-2 protein levels.14 The balance of reactive oxygen species (ROS) and antioxidant levels critically determines apoptosis in cancer cells, and overcoming the antioxidative defense systems by accelerating ROS production can promote apoptosis.15 Studies on autophagy induced by RES in cancer cells have also attracted a lot of attention.16 Autophagy can promote cancer cell adaptation and survival in response to various anticancer therapies by degradation of harmful components and damaged cellular organelles. Therefore, in this case, autophagy functions as a defensive role17,18 and inhibition of autophagy would enhance apoptosis in cancer cells. The factor that RES and PTX can modify different regulatory pathways involved in apoptosis suggests their combined use may yield a synergistic anticancer activity. However, little has been known by now whether RES influences PTX action through apoptosis and autophagy.
On the other hand, the low water solubility and therapeutic index of both RES and PTX make the simple combination of free drugs unsuccessful to achieve satisfactory combination settings in the body, which leads to their extremely limited clinical application.6,19 To this end, micelles entrapping PTX and RES have appeared as promising candidates that may improve their solubility and target them to tumors with optimal ratios for combination therapy via enhanced permeability and retention (EPR) effect due to the pathophysiological properties of solid tumors, such as hypervascularity, leaky vascular architecture, and the absence of effective lymphatic drainage.20
Therefore, in the present study, we constructed PTX and RES co-loaded nanoparticles based on amphiphilic methoxy poly (ethylene glycol)-b-polylactide (mPEG-b-PLA) block copolymers as drug carriers and evaluated their in vitro cytotoxicity and in vivo anti-tumor efficacy. The objective of this study was to elucidate the possible mechanism underlying the superior and selective anti-tumor efficacy of the combination therapy against single-drug systems from the following two aspects: ROS-mediated apoptosis and autophagy.
2. Materials and methods
2.1 Materials, cell lines, and animals
Resveratrol (RES) was purchased from Sigma-Aldrich (St. Louis, Mo, USA). Paclitaxel (PTX) was obtained from Zhejiang HISUN Pharmaceutical Co., Ltd. D,L-Lactide (DLLA) was supplied by DaiGang Biotechnology Co. Ltd. (Jinan, China). Monomethoxy poly(ethylene glycol) (mPEG, number average molecular weight is 2000 Da) purchased from Fluka (Steinheim, Germany) was dried by dissolution in anhydrous toluene followed by azeotropic distillation under N2 before use. Stannous octoate, Sn(Oct)2 was provided by Sigma Chemical (Steinheim, Germany). 2′,7′-Dichlorodihydrofluorescein diacetate was purchased from Sigma-Aldrich (St. Louis, Mo, USA). Methanol and acetonitrile for HPLC analysis was obtained from Merck (Darmstadt, Germany). Methyl thiazolyl tetrazolium (MTT) and dimethyl sulfoxide (DMSO) were purchased from Sigma (USA). BCA protein assay kit, β-actin antibody and Annexin V-FITC Apoptosis Kit were purchased from MultiSciences Biotech (China). Rabbit monoclonal anti-LC3 antibody and rabbit monoclonal BCl-2 antibody were obtained from Cell Signaling technology (USA). IRDye 680RD Goat anti-mouse and IRDye 800CW Goat anti-Rabbit secondary antibodies were obtained from LI-COR Biosciences (USA). Fetal bovine serum (FBS), RPMI-1640 medium (RPMI), and trypsin–EDTA (0.5% trypsin, 5.3 mM EDTA tetrasodium) were purchased from Ji Nuo Biotechnology Company (Hangzhou, China). All of other reagents and chemicals were analytical grade and were used without further purification.The PTX-resistant human lung adenocarcinoma epithelial (A549/T) cell line, normal human hepatic (L02) cell and normal human kidney (HK-2) cell line were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The mice sarcoma 180 (S180) cells were obtained from KeyGen Biotechnology Co., LTD (Nanjing, China). The cell lines were cultured in RPMI-1640 medium containing 10% FBS and 1% penicillin–streptomycin at 37 °C, 5% CO2 and humidified atmosphere.
Male ICR mice (4–6 weeks old, weighing 20 ± 2 g) were purchased from the Experimental Animal Center, Zhejiang Chinese Medical University (Hangzhou, China) and kept under SPF conditions. The animals were given daily fresh diet with free access to water and acclimatized for 6 days prior to the experiments. The animal experiments were conducted according to the Regulation on Experimental Animals of Zhejiang University.
2.2 Synthesis and characterization of mPEG-b-PLA block copolymers
In a flame-dried and nitrogen-purged flask, 1 g mPEG (Mn = 2000), 1 g DLLA, 5 ml toluene and 5 mg Sn(Oct)2 were added under nitrogen stream, and the sealed flask was maintained at 130 °C for 21 h under stirring. The synthesized polymer was recovered by dissolving in dichloromethane followed by precipitation in ice-cooled diethylether. The resultant precipitation was filtered and dried at room temperature in vacuum. After dissolving in CDCl3, the average molecular weight of block copolymers were determined by 1H NMR (500 MHz, Varian USA). The molecular weight distributions (PDI = Mw/Mn) were determined by gel permeation chromatography equipped with a Waters 515 HPLC pump, a Waters Styrage™ HT3 GPC column (300 mm in length and 7.8 mm in diameter), and a Waters 2410 refractive index detector. THF was used as a solvent, with a flow rate of 1.5 ml min−1 at 40 °C. Monodispersed polystyrene standards with a molecular weight range of 1310 to 5.51 × 104 from Waters Co. were used to generate the calibration curve.2.3 Preparation and characterization of polymeric micelles
Both blank and drug-loaded polymeric micelles (M-PTX, M-RES and M-PR) were prepared by thin film method.21 Briefly, 4 mg PTX or 4 mg RES and 50 mg mPEG-b-PLA were dissolved in 2 ml acetonitrile in a round-bottom flask. The solvent was evaporated by rotary evaporation at 50 °C for 30 min to obtain a solid PTX or RES/copolymer matrix. The resultant thin film was hydrated with 2 ml normal saline at 50 °C and vortexed for 5 min to obtain a micelle solution, which was then filtered through a 0.22 μm filtration membrane to remove the unincorporated PTX or RES. Finally, PTX-loaded micelles (M-PTX) or RES-loaded micelles (M-RES) were collected by freeze-drying. For the preparation of dual-drug loaded polymeric micelles, 1.25 mg PTX and 2.50 mg RES (M-PR (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)), 0.45 mg PTX and 3.6 mg RES (M-PR (1:8)) or 0.36 mg PTX and 3.6 mg RES (M-PR (1:10)) are dissolved with 50 mg mPEG-b-PLA in 2 ml acetonitrile followed with the same process described above. The blank PEG-b-PLA micelles were prepared in the same way except that PTX or RES was eliminated. All samples' solutions for in vitro cytotoxicity and in vivo anti-tumor efficacy evaluation were prepared by redissolving various micelles in sterile injection water and were filtered with the 0.22 μm sterile filtration membrane again before administration.
:2)), 0.45 mg PTX and 3.6 mg RES (M-PR (1:8)) or 0.36 mg PTX and 3.6 mg RES (M-PR (1:10)) are dissolved with 50 mg mPEG-b-PLA in 2 ml acetonitrile followed with the same process described above. The blank PEG-b-PLA micelles were prepared in the same way except that PTX or RES was eliminated. All samples' solutions for in vitro cytotoxicity and in vivo anti-tumor efficacy evaluation were prepared by redissolving various micelles in sterile injection water and were filtered with the 0.22 μm sterile filtration membrane again before administration.
The particle size and polydispersity index (PDI) were determined in double distilled water by dynamic light scattering (DLS, Nano-S90, Malvern, UK). The morphology of polymeric micelles was observed using a transmission electron microscope (TEM, JEM-1230, JEOL Ltd, Tokyo, Japan). A drop of sample solution (10 mg ml−1) was placed onto a 300 mesh copper grid coated with carbon without a staining agent. After 5 min, the grid was tapped with a filter paper to remove surface water. The grid was dried at room temperature and then observed by TEM.
Drug loading content (LC) and encapsulation efficiency (EE) of PTX or RES were determined by RP-HPLC with a UV detector (Agilent Technologies Inc. Cotati, CA, USA). For PTX, 100 μl PTX-loaded micelle solution was first dissolved in 1 ml acetonitrile followed by sonication and then diluted to 5 ml with the mobile phase. The analysis was performed on ODS C18 column (Diamonsil, 5 μm, 150 × 4.6 mm) and the wavelength was set at 227 nm. The mobile phase was a mixture of water, methanol and acetonitrile in the volume ration of 35:30:35. The elution rate was 1.0 ml min−1. For RES, 100 μl RES-loaded micelle solution was sonicated in 1 ml acetonitrile and diluted to 5 ml with pH 7.4 PBS. The analysis was performed on the same ODS column with the detection wavelength at 306 nm. The mobile phase was consisted of acetonitrile and 0.3% acetic acid solution (30:70, v/v). The flow rate was also 1.0 ml min−1. Each experiment was carried out in triplicate, and mean values ± SD deviations were calculated using the following formulas:
 | (1) |
 | (2) |
To examine the stability of drug-loaded micelles under physiological conditions, a solution of M-PR (1:8) was added to murine plasma at a final PTX concentration of 100 μg ml−1, and the sample was incubated at 37 °C. Aliquots were removed at 0, 1, 2, 4, 6, 10 and 24 h followed by analysis with RP-HPLC. PTX and RES retention was achieved from the weight ratio between the drug retained within micelles and that added at 0 h.
2.4 In vitro drug release study
The in vitro release behavior of PTX or RES from M-PTX, M-RES and M-PR, was monitored in PBS at pH 5.5, 6.5 and 7.4 by dialysis method. Briefly, 1 ml formulations with a known quantity (corresponding to 150 μg ml−1 of PTX and 1.5 mg ml−1 of RES) was introduced into a dialysis bag (MWCO: 8000–14000 Da) and the end-sealed dialysis bag was submerged fully into 20 ml PBS under the corresponding pH at 37 °C with stirring at 100 rpm for 72 h. At appropriate time intervals (0.5, 2, 4, 6, 8, 24, 48 and 72 h), 2 ml of the release medium was collected and replaced by the equal volume of fresh medium. The amount of PTX and RES were determined respectively by HPLC and percent cumulative release was calculated using the following formula:
 | (3) |
Cn is the sample concentration at Tn, V is the total volume of release medium, Vi is the sampling volume at Ti, Ci is the sample concentration at Ti (both V0 and C0 are equal to zero), and LC% is the percentage of drug loading.
2.5 Cell cytotoxicity assay
The MTT assay was employed to determine the in vitro cytotoxicity of the drug-loaded micelles against A549/T cells, S180 cells, L02 cells and HK-2 cells. Briefly, A549/T, L02 or HK-2 cells were seeded at a density of 5000 cells per well while S180 cells 10000 cells per well in 96-well culture plates and grown for 24 h under the condition of 5% CO2 at 37 °C. The medium was then added with various concentrations of different formulations and free drugs for 72 h at 37 °C. Then, 32 μl of MTT solution (5 mg ml−1 in PBS) was added into each well followed by 4 h incubation at 37 °C in darkness, after which, the medium was removed and 200 μl DMSO was added. Measurement was performed using Multiskan MK3 (Thermo, USA) at 570 nm. Cell viability of the control group was considered as 100%. Relative viability was calculated by comparing the absorbance intensity of groups treated with different drug-loaded micelles with the control group. The drug concentration of inhibition of 50% cell growth (IC50) was calculated by SPSS 18.0. All experiments were in triplicate, the results are given as mean ± standard deviation.
To estimate synergy, additivity, or antagonism of combination treatments, combination index (CI) values were determined using the method of Chou and Talaly.22 The CI values were calculated as follows:
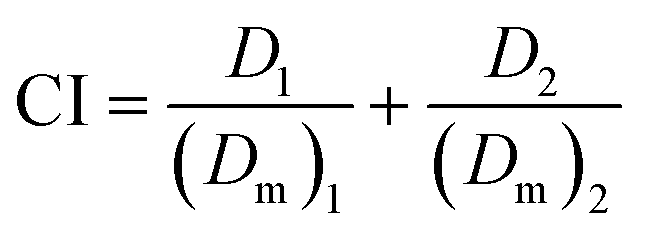 | (4) |
(Dm)1 and (Dm)2 represent the IC50 of treatments 1 and 2 applied separately, while D1 and D2 are IC50 of treatments 1 and 2 applied as a combination.
2.6 Apoptosis inducing effect
Apoptotic cell death was determined by staining with Annexin V-FITC apoptosis detection kit (Beyotime, Jiangsu, China) according to the manufacturer's protocol using the flow cytometer. Briefly, A549/T and S180 cells were seeded in 6-well cell culture plates at a density of 2 × 105 cells per well in 2 ml of growth medium. After 24 h, the cells were treated with different formulations for 48 h under the condition of 5% CO2 at 37 °C. The concentration of PTX in M-PTX, M-PR (1:2) and M-PR (1:8) was 5 μg ml−1 and the concentration of RES in M-RES(L) and M-PR (1:2) was 10 μg ml−1 while in M-RES(H) and M-PR (1:8) was 40 μg ml−1. Control experiments were performed by adding normal saline. Then the cells were harvested and suspended in 500 μl of provided binding buffer, after which, 5 μl of Annexin V-FITC and 10 μl PI was added. Then the mixture was incubated for 5 min at room temperature in the dark. The cells were immediately analyzed by a FACScan flow cytometry with the events collected 1 × 104. Each assay was repeated in triplicate.
2.7 Determination of intracellular ROS levels
Intracellular ROS generation in A549/T, L02 and HK-2 cells was examined by cytometry using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). DCFH-DA is a lipid permeable nonfluorescent compound and is oxidized by intracellular ROS to form the lipid impermeable and fluorescent compound DCF. Briefly, A549/T cells were seeded in 6-well cell culture plates at a density of 2 × 105 cells per well in 2 ml of growth medium. After 24 h, the cells were treated with different formulations for 12 h under the condition of 5% CO2 at 37 °C. The concentration of PTX in M-PTX, M-PR (1:2) and M-PR (1:8) was 25 μg ml−1 and the concentration of RES in M-RES(L) and M-PR (1:2) was 50 μg ml−1 while in M-RES(H) and M-PR (1:8) was 200 μg ml−1. Control experiments were performed by adding normal saline. After treatment, the cells were washed with PBS and then incubated with 10 μM DCFH-DA in RPMI-1640 medium at 37 °C for 1 h in the dark. Finally, the cells were washed three times with PBS, trypsinized (0.05% trypsin) and then resuspended in 500 μl of PBS. ROS generation of these cells was determined by a FACscan flow cytometry using 488 nm for excitation and 525 nm for emission.
2.8 Western blot analysis
Drug-loaded micelles treated A549/T and S180 cells were examined by western blot analysis for BCl-2 and autophagy marker Light Chain 3 (LC3) levels in the whole-cell extracts. The whole-cell extracts were prepared by lysing cells in lysis buffer containing a protease inhibitor cocktail after having been treated with different formulations for 48 h. The total protein concentration was determined by BCA protein assay kit. Protein samples were separated with 8–15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). After electrophoresis, the proteins were electrotransferred to 0.2 μm poly(vinylidene fluoride) membranes (PVDF, Millipore), blocked with 5% nonfat milk for 1 h, and probed with BCl-2, LC3 and β-actin antibodies at 4 °C overnight. The blots were then washed, exposed to IRDye-680RD Goat anti-mouse and IRDye 800CW Goat anti-Rabbit secondary antibodies (1:10000) for 1 h, and finally examined by the Odyssey® scanner (LI-COR, USA).
2.9 In vivo anti-tumor activity and histopathological examination
The in vivo inhibitory efficacy of drug-loaded micelles was evaluated against S180 solid tumor bearing mice model. After S180 cells being subcutaneously implanted into the armpits of the mice, the tumor bearing mice were randomly divided into five groups (6 mice per group): control (normal saline), M-PTX, M-RES, M-PR (1:8), M-PTX + M-RES, with PTX dose of 20 mg kg−1 and RES dose of 160 mg kg−1. The dose schedule started when the tumor volume was about 100–200 mm3 (5 days after implantation) and treatments were given four times through tail vein injection once every 2 days. The body weight of each mouse, length, and width of tumor were determined every alternate day. The tumor volume was calculated as the following: V = [length × (width)2]/2. On the day 8, the animals were sacrificed by cervical dislocation, and the tumor mass was harvested, weighted, photographed, and processed for histopathological examination. The inhibitory of rate of tumor (IRT%) was calculated according to the formula:| IRT% = (Wcontrol − WTreated)/Wcontrol × 100% | (5) |
Xenograft tumors of various treatment groups were fixed with 4% formaldehyde for 48 h and embedded in paraffin. Each section was cut into 4 μm, processed for routine hematoxylin and eosin (H&E) staining, and then visualized under fluorescent microscope. For analysis of tumor vasculature, CD31 antibody (1:50; Abcam, Cambridge, UK) was used for microvessel staining, and sections were incubated with Ki67 antibody (1:200; Abcam, Cambridge, UK) for cell proliferation analysis.
2.10 Statistical analysis
Data were expressed as mean ± SD and statistically analyzed using the one way analysis of variance (ANOVA). A significant difference was considered as p < 0.05, and very significant difference was regarded as p < 0.01.3. Results and discussion
3.1 Synthesis and characterization of block copolymers
mPEG-b-PLA copolymer was ring-opening polymerized from DLLA in the presence of mPEG as an macromolecular initiator. The polymer structure was confirmed by 1H NMR analysis in CDCl3 and the spectrum is shown in Fig. 1. The peaks at 1.65 and 5.20 ppm belong to methine (–CH) and methyl protons (–CH3) of PLA segments, respectively, while the methylene protons (–CH2–) of PEG segment appear at 3.65 ppm. This demonstrates successful synthesis of mPEG-b-PLA copolymer with high purity. From the ratio of peak area at 5.20 and 3.65 ppm, the number average molecular weight of the resultant copolymer was calculated as 3963.6. GPC result shows that the number-average molecular weight (Mn) of mPEG-PLA was 4173, which was close to the NMR result, with a narrow polydispersity of 1.137. The fEO (volume fractions of the mPEG blocks) was 49.56% which was converted from the measured mass fractions by using homopolymer melt densities: 1.13 and 1.09 g cm−3 for PEG and PLA, respectively.23 As previously reported, mPEG-b-PLA copolymers with hydrophilic PEG fractions (fEO) in the ranges of 42–50 % or even over 50% usually form worm micelles and spherical micelles, respectively.23 Therefore, it was predicted that the resultant mPEG-b-PLA copolymers could self-assemble in the form of micelles in an aqueous solution with the capability to load hydrophobic drugs. | ||
| Fig. 1 1H NMR spectrum of mPEG-b-PLA in CDCl3. | ||
3.2 Drug loading of mPEG-b-PLA micelles
Various drug-loaded micelles were prepared by thin film method. As shown in Table 1, the total drug loading content (PTX, RES, PTX + RES) in single or dual drug-loaded micelles was all set as about 8% and the loading ratio of PTX to RES could be adjusted from 1:2 to 1:10. Moreover, the encapsulation efficiencies of PTX or RES in different formulations were all as high as above 95% regardless of PTX and RES ratio. The mean particle size of drug-loaded micelles was a little larger than that of blank micelles (Table 1). Fig. 2 shows the typical TEM image and size distribution of M-PR (1:8). M-PTX, M-RES and the other M-PRs displayed the similar morphology.
| Preparations | Particle Size (nm) | PDI | Drug Loading Content (%) | Drug Encapsulation Efficiency (%) | ||
|---|---|---|---|---|---|---|
| PTX | Res | PTX | Res | |||
| a Data are presented as mean ± SD (n = 3). | ||||||
| Blank micelles | 20.51 ± 1.57 | 0.12 ± 0.02 | — | — | — | — |
| M-PTX | 21.70 ± 1.44 | 0.17 ± 0.03 | 7.96 ± 0.06 | — | 98.72 ± 1.23 | — |
| M-RES | 23.13 ± 1.53 | 0.21 ± 0.02 | — | 7.93 ± 0.07 | — | 96.96 ± 2.57 |
| M-PR(1:2) |
22.30 ± 1.54 | 0.21 ± 0.01 | 2.47 ± 0.12 | 5.13 ± 0.06 | 98.47 ± 0.64 | 98.55 ± 1.24 |
| M-PR(1:8) |
21.87 ± 1.46 | 0.13 ± 0.04 | 1.03 ± 0.06 | 7.99 ± 0.07 | 98.15 ± 1.72 | 96.00 ± 2.38 |
| M-PR(1:10) |
22.26 ± 1.52 | 0.19 ± 0.02 | 0.68 ± 0.03 | 7.11 ± 0.05 | 97.43 ± 0.89 | 95.39 ± 1.75 |
 | ||
| Fig. 2 Characterization of dual drug-loaded micelles (M-PR (1:8)). (A) Typical TEM image of M-PR (1:8). Scale bar = 200 nm. (B) Size distribution of M-PR (1:8) characterized by DLS. | ||
Fig. 3 shows the release behaviors of PTX and RES from the drug-loaded micelles at pH 5.5, pH 6.5 and pH 7.4. The pH selection depended on the application environments, including the physiological fluids (pH 7.4), tumor tissue (pH 6.5) and endosomal compartments (pH 5.5). Interestingly, we found that the release of PTX and RES was almost synchronized in single-drug micelles, however, in the dual-drug loaded formulations, the release profile of RES was increased a little while that of PTX was seriously retarded with only 65% release approximately in 72 h. This notable discrepancy in release rate of PTX and RES from dual drug-loaded micelles might be attributed to the different interaction strength between drug and hydrophobic PLA blocks in mPEG-b-PLA micelles. In the study, we found that, if the EE was requested more than 95%, the maximum RES loading was about 8% while that of PTX could reached 20%. This phenomenon suggested that PTX was more compatible with PLA than RES according to like dissolves like theory, which means any substance that has the same polarity (either polar or non polar) dissolve into each other.24,25 It has been reported that PTX is a highly hydrophobic drug that is poorly soluble in water (0.5–35 μM),26 while the solubility of RES in water (175.25 μM)27 is much higher. As a result, during the encapsulation procedure of both PTX and RES, PTX, as the more hydrophobic compound, was interacted with PLA more intensely than RES, which induced different release behaviors. The prolonged release profile of PTX compared to RES would reach much greater synergism of the binary combination therapy due to sequence-dependence of the combined drug action, that is, administration of RES before PTX may sensitize the cancer cells to PTX-induced apoptosis.28 Since the aqueous solubility of both PTX and RES was not obviously changed at different pH values, the release profile under different pH conditions was almost the same.
 | ||
| Fig. 3 In vitro release profiles of PTX and RES from M-PTX, M-RES and M-PR in various release media at different pH values. The release media was set as at (A) pH 7.4, (B) pH 6.5, and (C) pH 5.5. | ||
We further investigated the stability of the drug loaded micelles (M-PR (1:8)) in murine plasma at 37 °C to simulate normal physiological blood conditions. As shown in Fig. 4, more than 90% of both PTX and RES remained after 24 h incubation, indicating the satisfactory stability of M-PR (1:8) in plasma.
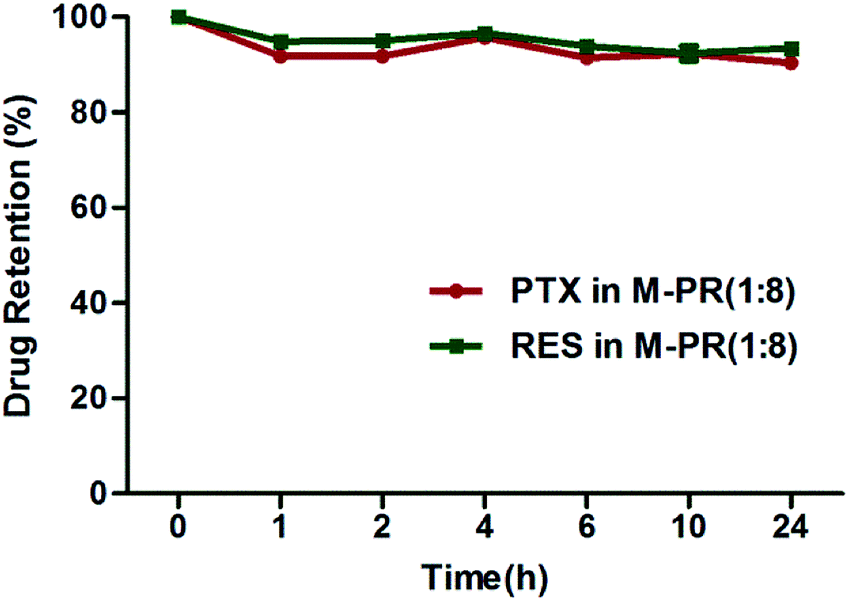 | ||
| Fig. 4 The stability of M-PR (1:8) after 1, 2, 4, 6, 8, 24 h incubation in the murine plasma at 37 °C. (n = 3). | ||
3.3 Cell cytotoxicity assay
The particle size within the 10–100 nm enables the drug-loaded micelles to be transported to tumor tissue without rapidly being eliminated by the kidney or without inhibited by the nascent vasculature of tumors.29 However, it has demonstrated that small particles show a widespread organ distribution including liver and kidney30 and thus cause undesirable systemic toxicity. Therefore, in vitro cytotoxicity assay in both tumor cells as well as normal liver and kidney cells is necessary.Reduction in IC50 values of chemotherapeutic agents should be one of the major therapeutic benefits of this combination therapy. By MTT assays, the IC50 of M-PTX and M-RES was determined as 1.427 μg ml−1 and 8.525 μg ml−1 in A549/T cells, respectively. As shown in Fig. 5A, once PTX and RES were co-loaded into micelles, the IC50 of PTX was gradually decreased with the increase of RES content until the ratio of PTX to RES reached 1:10. The corresponding CI values summarized in Table 2 indicated that the combination of PTX and RES at ratios of 1:2 and 1:8 could achieve a synergistic effect, however, it turned to antagonism at a ratio of 1:10 in A549/T. As for S180 cells, PTX is quite insensitive with high IC50 value (424.571 μg ml−1). Co-loading RES with PTX at 1:8 weight ratio in the micelles significantly decreased the IC50 of PTX to 0.701 μg ml−1 with synergism effect. Therefore, M-PR (1:8) was optimized in the antitumor effects study in vivo. On the other hand, the IC50 values of free PTX and RES against A549/T and S180 cells were also calculated to investigate whether drug-loaded nanoparticles have an advantage over the free drugs. The IC50 of free PTX and RES was 9.667 ± 1.192 μg ml−1 and 37.049 ± 0.195 μg ml−1 respectively in A549/T cells, while the IC50 of free RES was 31.441 ± 1.082 μg ml−1 in S180 cells. However, the maximum solubility of free PTX in RPMI-1640 medium is 200 μg ml−1, under which the cell viability was still above 70%. Therefore, the IC50 of free PTX in S180 cells could not be determined. According to these results, A549/T and S180 cells became more sensitive to PTX and RES with the help of the polymer carrier.
 | ||
| Fig. 5 Cell cytotoxicity of different formulations towards A549/T, S180 and normal cells at 72 h. (A) IC50 of single drug-loaded micelles (M-PTX and M-RES) or dual drug-loaded micelles in A549/T (M-PR (1:2); M-PR (1:8); M-PR (1:10)) in A549/T cells (n = 3). (a), p < 0.01, vs. M-PTX; (b), p < 0.01, vs. M-PR(1:8). (B) Cell viability (%) of A549/T, S180, L02 and HK-2 cells after applying M-PTX (n = 3). (C) Cell viability (%) of A549/T, S180, L02 and HK-2 cells after applying M-RES (n = 3). (D) Cell viability (%) of A549/T, S180, L02 and HK-2 cells after applying M-PR (1:8) (n = 3). | ||
| Preparations | A549/T | S180 | ||||
|---|---|---|---|---|---|---|
| IC50 ± SD (μg ml−1) | CI | IC50 ± SD (μg ml−1) | CI | |||
| PTX | RES | PTX | RES | |||
| a Data are presented as mean ± SD (n = 3). CI < 0.1, very strong synergism; CI = 0.1–0.3, strong synergism; CI = 0.3–0.7, synergism; CI = 0.7–0.85, moderate synergism; CI = 0.85–0.90, light synergism; CI = 0.90–1.10, nearly additive; CI = 1.10–1.20, slight antagonism; CI = 1.20–1.45, moderate antagonism; CI = 1.45–3.3, antagonism; CI = 3.3–10, strong antagonism; and, CI > 10, very strong antagonism. | ||||||
| M-PTX | 1.427 ± 0.216 | — | — | 424.571 ± 9.660 | — | — |
| M-RES | — | 8.525 ± 0.033 | — | — | 7.435 ± 0.340 | — |
| M-PR (1:2) |
0.752 ± 0.042 | 1.339 ± 0.075 | 0.68 | 3.505 ± 0.189 | 7.010 ± 0.378 | 0.95 |
| M-PR (1:8) |
0.464 ± 0.010 | 3.374 ± 0.071 | 0.72 | 0.701 ± 0.085 | 5.607 ± 0.681 | 0.76 |
| M-PR (1:10) |
0.622 ± 0.074 | 6.139 ± 0.727 | 1.16 | 2.577 ± 0.164 | 27.054 ± 1.719 | 3.64 |
To verify whether the combination of PTX and RES influenced the growth of normal cells, inhibitory effects of M-PTX, M-RES and M-PR (1:8) on L02 and HK-2 cells was also evaluated and compared with that on A549/T and S180 cells (Fig. 5B–D). Micelles loaded with single PTX performed considerable toxicity towards normal hepatic and kidney cells in accord with previous research, that is, the cell viability was all below 50% under the PTX concentration from 0.31-5 μg ml−1 in L02 (from 45.41% to 42.31%) and HK-2 (from 49.87% to 46.73%) cells. However, in the treatment of M-PR (1:8), as PTX concentration changed in the range of 0.31–5 μg ml−1, the cell viability was decreased from 53.81 % to 6.71 % in A549/T and from 75.41 % to 7.60 % in S180, while it was still above 50% at the concentration of 5 μg ml−1 in both L02 (59.99%) and HK-2 (73.37%) cells. Interestingly, the variation trend was similar in the treatment of M-RES under the same RES concentration range. With the RES concentration ranged from 2.5–40 μg ml−1, the cell viability was decreased from 75.94% to 17.70% in A549/T and from 78.96% to 8.52% in S180, while in L02 and HK-2, it was from 100% to 58.18% and from 94.67% to 48.01%. This result indicates that in virtue of RES, M-PR (1:8) can selectively enhance the cytotoxicity to tumor cells but without much adverse effect towards normal cells.
3.4 Apoptosis inducing effect
Induction of programmed cell death including apoptosis has been regarded as a very important strategy to eliminate cancer cells in the chemotherapy. Therefore, the regulation of RES on the cytotoxicity of PTX toward A549/T and S180 cells was also evaluated by quantification of apoptotic cells. As shown in Fig. 6A, M-PTX of 5 μg ml−1 and M-RES(L) of 10 μg ml−1 treatment resulted in 42.08% (11.72% + 30.36%) and 37.26% (10.63% + 26.63%) of apoptosis, respectively, which grew to 44.05 (9.12% + 34.93%) when exposed to M-PR (1:2). Similarly, M-RES(H) of 40 μg ml−1 treatment resulted in 47.03% (9.12% + 34.93%) of apoptosis, while the combination therapy of M-PR (1:8) induced further more apoptosis up to 61.29% (9.12% + 34.93%). Comparing M-PR (1:2) treatment with M-PR (1:8), we drew the conclusion that RES raised PTX-induced apoptosis percentage in a dose-dependent manner with PTX:RES ratios ranging from 1:2 to 1:8 in A549/T cells. The same character of M-PR could be observed in S180 cells (Fig. 6B) with the most significant apoptosis inducing effect (46.49% (11.43% + 35.06%)) for M-PR (1:8). These results were consistent with the above-mentioned cytotoxicity results, indicating that the increased sensitivity of A549/T and S180 cells to PTX is possibly attributed to the enhanced PTX-induced apoptosis in the presence of RES.
 | ||
| Fig. 6 Apoptotic rates of tumor cells treated with blank micelles, M-PTX, M-RES(L), M-RES(H), M-PR(1:2), and M-PR (1:8). Apoptotic rates of (A) A549/T cells and (B) S180 cell treated with different formulations. | ||
3.5 Intracellular ROS levels determination
A moderate increase in ROS can promote cell proliferation and differentiation,31 whereas excessive amounts of ROS will cause oxidative damage to lipids, proteins and DNA.32 We found a combination of PTX and RES (1:8) to be pro-apoptotic for A549/T and S180 while normal cells were resistant according to the result of cell cytotoxicity. The promoted apoptosis in A549/T and S180 cells can be further validated in apoptosis inducing assays. Therefore, we examined whether alteration of the intracellular ROS accumulation was contributed from PTX and RES ratios. As shown in Fig. 7A, we confirmed that the intracellular ROS accumulation of M-PTX treatment group was two-fold increased in comparison with controls while that could increase 0.8 times and 18.3 times for M-RES treatment at a low concentration and high concentration, respectively. And M-PR (1:8) could improve intracellular ROS accumulation most significantly, 27.8 times as likely to controls in A549/T cells, which indicates that PTX and RES combination exerted synergistic effect on the induction of intracellular ROS levels in cancer cells. However, there was no significant change in the intracellular ROS accumulation both in normal L02 and HK-2 cells when treated with different formulations. This phenomenon was consistent with the selective cytotoxicity to A549/T and S180 cancer cells. Therefore, it was inferred that PTX and RES co-loaded nanoparticles can induce more intracellular ROS of cancer cells, which in turn selectively enhance the cytotoxicity to tumor cells. This phenomenon can be briefly illustrated in Fig. 7B. Normal cells can tolerate a certain level of exogenous ROS stress owing to their ‘reserve’ antioxidant capacity, which can be mobilized to prevent the ROS level from reaching the cell-death threshold.31 Compared with their normal counterparts, many types of cancer cell have increased levels of ROS,32 which is associated with abnormal cancer cell growth and a disruption of redox homeostasis due either to an elevation of ROS production or to a decline of ROS-scavenging capacity, a condition known as oxidative stress.33 As a result, cancer cells with increased oxidative stress are likely to be more vulnerable by further ROS induced by exogenous agents.34 Namely, a further increase in cancer cells (red bar) using exogenous ROS-modulating agents is likely to cause elevation of ROS above the threshold level, leading to cell death. This might constitute a biochemical basis to selectively killing cancer cells using ROS-mediated mechanisms.
 | ||
| Fig. 7 Apoptosis inducing effect of various formulations through enhanced intracellular ROS levels. (A) Intracellular ROS generation by the treatment of blank micelles, M-PTX, M-RES(L), M-RES(H), M-PR (1:2), and M-PR (1:8) in L02, HK-2 and A549/T cells (n = 3). *p < 0.05. (B) Biochemical basis to selectively killing cancer cells using ROS-mediated mechanisms. | ||
3.6 Modulation of BCl-2 and LC3 protein levels by drug-loaded micelles
Apoptosis is a process of life involving a variety of genes including the anti-apoptosis gene BCl-2, whose expression is reported to localize at mitochondria and stabilize mitochondrial functions, thereby suppressing the release of proapoptotic effector molecules.35 Recent studies reported that the microtubule-targeting agents could inactivate the BCl-2 family proteins in cancer cells.36 In this study, we examined how different formulations modulated the expression of BCl-2. As shown in Fig. 8A and B, treatment with PTX or RES at low concentration alone or in combination did not affect the levels of BCl-2 significantly. However, the presence of RES at high concentration alone can reduce the anti-apoptotic BCl-2 protein levels, and co-presence of PTX can reduce the BCl-2 expression further both in A549/T and S180 cells. The results above supports the notion that enhanced cytotoxicity by RES is partly attributed to its inhibition of expression of BCl-2 in a dose-dependent manner. | ||
| Fig. 8 Modulation of Bcl-2 and LC3 protein levels by different formulations towards tumor cells. Apoptosis inducing effect of drug-loaded micelles through reducing anti-apoptotic Bcl-2 protein and autophagy inhibition in (A) A549/T cells and. (B) S180 cells. | ||
Next we evaluated whether cancer cells treated with different formulations also underwent autophagy. Autophagy is initiated in response to cellular stress by autophagosome formation, which requires the conversion of microtubule associated protein 1 light chain 3 (LC3-I) to the autophagosome associated LC3-II.37 Therefore, we determined the levels of LC3-I and LC3-II upon different treatments by western blot analysis in S180 and A549/T cells. It was observed that a high concentration of RES and the combination of PTX and RES at a ratio of 1:8 can reduce LC3-II protein level and thus suppressed autophagic pathway, which would enhance PTX-induced apoptosis in A549/T and S180 cells as shown in Fig. 6. This result is consistent with the previous report that inhibiting autophagy via RES promoted temozolomide-induced apoptosis in brain tumors.38
To sum up, the most significant cytotoxicity of dual-drug micelles toward A549/T and S180 is further evidenced by the reduction of antiapoptotic BCl-2 and autophagy marker LC3 expression levels.
3.7 In vivo anti-tumor efficacy and histological analyses
The in vivo anti-tumor activity of different treatments was evaluated in mice bearing S180 tumors. On the 8th day, the mice treated with M-PTX showed abnormal physiological behaviors such as poor appetite and significant body weight loss. This severe adverse reaction may be resulted from the faster PTX release from M-PTX during the circulation than that from M-PR (1:8). Therefore, the experiment of anti-tumor efficacy evaluation lasted for 8 days. In the tumor growth profiles (Fig. 9A), the tumor volumes in the mice treated with M-PR (1:8) were lower than that of saline and other formulations. Meanwhile, the tumor inhibitory rate of M-PR (1:8) was 67.00 ± 9.52%, which was higher than that of M-PTX (48.74 ± 14.80%), M-RES (23.41 ± 14.79%) and M-PTX + M-RES (39.01 ± 15.06%) (Fig. 9B). Thereby, M-PR (1:8) could be most effective to inhibit the tumor growth among all the treatment groups. It should be noticed that single M-PTX was more effective than M-PTX + M-RES according to the tumor inhibitory rate. The major cause for this phenomenon is that simple combination of M-PTX and M-RES can't guarantee satisfactory combination settings in the body, that is, the ratio between PTX and RES would not be exactly 1:8 in the tumor cells. And the changed ratio between two drugs may induce antagonism in inhibiting the growth of tumor according to the result in cell cytotoxicity assay. Actually this result clarified the importance of co-encapsulating PTX and RES into one micelle at the optimized ratio.
 | ||
| Fig. 9 In vivo anti-tumor activity of drug-loaded micelles in S180 bearing mice and histological analyses towards the excised tumors. (A) The tumor growth inhibitory profiles treated with saline, M-PTX, M-Res, M-PTX + M-Res, and M-PR (1:8) (n = 6). (B) The growth inhibitory rate of different treatments in comparison with that of saline group (n = 6). (a), p < 0.05, vs. M-PR (1:8); (b), p < 0.01, vs. M-PR (1:8). (C) H&E stained tumor slices from mice on the 8th day after treatment with various formulations. Scale bar = 50 μm. | ||
To evaluate the apoptosis in the tumors treated with saline, M-PTX, M-RES, M-PTX + M-RES and M-PR (1:8), the tumors were excised from the mice of the experimental groups after treatment, and tumor slices stained by hematoxylin and eosin (H&E) were photographed via optical microscopy. As presented in Fig. 9C, it was demonstrated that apoptosis occurred in S180 tumor slices treated with various formulations. It was clear that cell nuclei apoptosis of M-PR (1:8) group was very severe. In contrast, M-PTX and M-RES treated tumors displayed a much lower necrotic level with a large amount of living tumor cells existing. The histological studies provide evidence that dual-drug micelles evoke definitive changes in the cytoarchitecture leading to tumor regression. Although single-drug systems also affect the tumor, the combination therapy leads to more obvious histopathological abnormalities, degenerations and lesions with negligible tumor residue, indicating severe tumor tissue damage.
The findings above have several implications. First, dual-drug systems exhibits a better antitumor effect than single-drug systems by an additive action of apoptosis inducing effect, suggesting the synergistic effect of PTX and RES in vivo. Furthermore, the superiority of M-PR (1:8) to M-PTX + M-RES validated that the dual drug in one micellar system with different release rates can facilitate tumor cell killing.
4. Conclusions
In summary, micelles loaded with PTX and RES were prepared by thin film method using mPEG-PLA block copolymers with a small size, high encapsulation and sequential drug release. In vitro cytotoxicity test, the combination strategy exhibited synergistic effect in A549/T and S180 cells while showing very limited toxicity towards normal cells, which could be illustrated from enhanced apoptosis and reduced protective autophagy. The apoptosis inducing effect of this dual-drug system was further evidenced by elevated amounts of intracellular ROS and reduction of anti-apoptotic BCl-2. In vivo, PTX and RES co-loaded micelles achieved the best antitumor effect in all treatment groups in S180 bearing mice. Moreover, the histological studies provided evidence that dual-drug micelles evoked the most significant changes in the cytoarchitecture leading to tumor regression. On the whole, the combination therapy of PTX and RES in one micelle system would provide a potential strategy to treat cancers by apoptosis inducing effect.Acknowledgements
This study was financially supported by the National Natural Science Funds for Excellent Young Scholar (81222047), Zhejiang Nature Science Foundation of China (Y2110478), the Fundamental Research Funds for the Central Universities (2014XZZX003-20) and the Ministry of Education Program for New Century Excellent Talents (NCET-11-0454).References
- R. Siegel, D. Naishadham and A. Jemal, Ca-Cancer J. Clin., 2012, 62, 10 CrossRef PubMed.
- P. Cao and Y. Bae, Future Oncol., 2012, 8, 1471 CrossRef CAS PubMed.
- A. Persidis, Nat. Biotechnol., 1999, 17, 94 CrossRef CAS PubMed.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044 CrossRef CAS PubMed.
- J. A. Baur and D. A. Sinclair, Nat. Rev. Drug Discovery, 2006, 5, 493 CrossRef CAS PubMed.
- A. K. Singla, A. Garg and D. Aggarwal, Int. J. Pharm., 2002, 235, 179 CrossRef CAS.
- S. J. Flatters and G. J. Bennett, Pain, 2004, 109, 150 CrossRef CAS PubMed.
- T. Kubota, Y. Uemura, M. Kobayashi and H. Taguchi, Anticancer Res., 2002, 23, 4039 Search PubMed.
- F. Quan, C. Pan, Q. Ma, S. Zhang and L. Yan, Biomed. Pharmacother., 2008, 62, 622 CrossRef CAS PubMed.
- J. Gusman, H. Malonne and G. Atassi, Carcinogenesis, 2001, 22, 1111 CrossRef CAS PubMed.
- J. K. Kundu and Y.-J. Surh, Cancer Lett., 2008, 269, 243 CrossRef CAS PubMed.
- A. Bishayee, Cancer Prev. Res., 2009, 2, 409 CrossRef CAS PubMed.
- M. E. l. Juan, U. Wenzel, H. Daniel and J. M. Planas, J. Agric. Food Chem., 2008, 56, 4813 CrossRef CAS PubMed.
- Y.-B. Ouyang and R. G. Giffard, Neurochem. Int., 2004, 45, 371 CrossRef CAS PubMed.
- J. Shao, X. Li, X. Lu, C. Jiang, Y. Hu, Q. Li, Y. You and Z. Fu, Colloids Surf., B, 2009, 72, 40 CrossRef CAS PubMed.
- J.-y. Wu, K.-w. Tsai, J.-j. Shee, Y.-z. Li, C.-h. Chen, J.-j. Chuang and Y.-w. Liu, Acta Pharmacol. Sin., 2010, 31, 81 CrossRef CAS PubMed.
- T. Hara, K. Nakamura, M. Matsui, A. Yamamoto, Y. Nakahara, R. Suzuki-Migishima, M. Yokoyama, K. Mishima, I. Saito and H. Okano, Nature, 2006, 441, 885 CrossRef CAS PubMed.
- K. Degenhardt, R. Mathew, B. Beaudoin, K. Bray, D. Anderson, G. Chen, C. Mukherjee, Y. Shi, C. Gelinas and Y. Fan, Cancer Cell, 2006, 10, 51 CrossRef CAS PubMed.
- F. Caruso, J. Tanski, A. Villegas-Estrada and M. Rossi, J. Agric. Food Chem., 2004, 52, 7279 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271 CrossRef CAS.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621 CrossRef CAS PubMed.
- F. Ahmed and D. E. Discher, J. Controlled Release, 2004, 96, 37 CrossRef CAS PubMed.
- K. A. Barnes, A. Karim, J. F. Douglas, A. I. Nakatani, H. Gruell and E. J. Amis, Macromolecules, 2000, 33, 4177–4185 CrossRef CAS.
- H. Minjia, T. Chao, Z. Qunfang and J. Guibin, J. Chromatogr. A, 2004, 1048, 257–262 CrossRef PubMed.
- D.-B. Chen, T.-z. Yang, W.-L. Lu and Q. Zhang, Chem. Pharm. Bull., 2001, 49, 1444–1447 CrossRef CAS.
- V. Filip, M. Plocková, J. Šmidrkal, Z. Špičková, K. Melzoch and Š. Schmidt, Food Chem., 2003, 83, 585–593 CrossRef CAS.
- M. U. Nessa, P. Beale, C. Chan, J. Q. Yu and F. Huq, Anticancer Res., 2012, 32, 53 CAS.
- J. D. Heidel and M. E. Davis, Pharmaceut. Res., 2011, 28, 187 CrossRef CAS PubMed.
- W. H. De Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. Sips and R. E. Geertsma, Biomaterials, 2008, 29, 1912 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579 CrossRef CAS PubMed.
- S. Kawanishi, Y. Hiraku, S. Pinlaor and N. Ma, Biol. Chem., 2006, 387, 365 CrossRef CAS PubMed.
- S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1 CrossRef CAS.
- H. Pelicano, D. Carney and P. Huang, Drug Resist. Updates, 2004, 7, 97 CrossRef CAS PubMed.
- I. Tinhofer, D. Bernhard, M. Senfter, G. Anether, M. Loeffler, G. Kroemer, R. Kofler, A. Csordas and R. Greil, FASEB J., 2001, 15, 1613 CAS.
- M. Fukui and B. T. Zhu, Mol. Carcinog., 2009, 48, 66 CrossRef CAS PubMed.
- V. Prabhu, P. Srivastava, N. Yadav, M. Amadori, A. Schneider, A. Seshadri, J. Pitaressi, R. Scott, H. Zhang and S. Koochekpour, Mitochondrion, 2013, 13, 493 CrossRef CAS PubMed.
- C.-J. Lin, C.-C. Lee, Y.-L. Shih, T.-Y. Lin, S.-H. Wang, Y.-F. Lin and C.-M. Shih, Free Radical Bio. Med., 2012, 52, 377 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |