
Progress in the Suzuki polycondensation of fluorene monomers†
Abstract
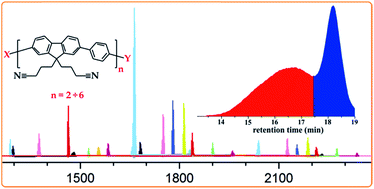
A step-growth AA/BB type Suzuki polycondensation approach has been applied to synthesize two high molecular weight polyfluorenes bearing nitrile-terminated short alkyl side chains and protected amino functionalities. The polymers were obtained reproducibly on the gram scale and as typical polyfluorene-based materials. Three different often used palladium catalysts and several reaction parameters were evaluated in order to optimize the conditions towards high molar masses. The issues encountered during the polymerizations led to a thorough investigation by in-depth mass spectrometric analysis of the oligomeric species obtained as side products. This resulted in some mechanistic insights showing ligand scrambling as the major side reaction. The decorated polyfluorenes reported here seem to be the best choice so far for post-polymerization modifications performed on high molecular weight conjugated polymers with lateral functional groups.
 Please wait while we load your content...
Please wait while we load your content...

