
Adsorption of CO, SO2, HCN, NH3, and H2CO on zigzag GaP nanotubes: a QM/MM study†
Pabitra Narayan Samanta and
Kalyan Kumar Das*
Department of Chemistry, Physical Chemistry Section, Jadavpur University, Kolkata 700 032, India. E-mail: kkdas@chemistry.jdvu.ac.in
First published on 23rd October 2014
Abstract
The interactions of the single-walled zigzag (5, 0), (6, 0), and (7, 0) GaP nanotubes (GaPNTs) with CO, SO2, HCN, NH3, and H2CO molecules are theoretically studied at the ONIOM(B3LYP/6-31G(d):UFF) level. A pyrene-like ring of the nanotube is chosen as an adsorption site in the high layer of the ONIOM calculations for the adsorption of a single molecule. Binding energy, Gibbs free energy change, density of states, and Mulliken charge transfer are computed to analyze the nature of the binding between GaPNT and the adsorbate molecule. The bindings of CO and H2CO towards GaPNTs are weaker than those of NH3, SO2, and HCN molecules. The strongest adsorption is found to be with NH3. The dispersion corrected functional (wB97XD) has been introduced to compare the results with those of the B3LYP functional.
1. Introduction
Since the discovery of carbon nanotubes (CNTs) by Iijima,1 efforts are being made for its development as materials such that it can be used as gas storage elements, as sensors in nanoelectronics, and as bio-compatible agents in medicine.2 The adsorptions of NO2 and NH3 gases on CNTs at room temperature have been found to change the conductivity of the nanotubes largely.3 Molecules like O2 and SO2 also interact with CNTs strongly.4,5 First-principles calculations on the interactions of NO2, O2, NH3, N2, H2O, and Ar with CNTs have been carried out by Zhao et al.6 Over the years, there is a strong zeal to improve the sensitivities of CNTs towards toxic gases like CO, H2S, and H2CO using different strategies. A substitutional doping with Si atom has been found to be very effective in increasing the reactivity of CNTs towards toxic gases.7,8 Silicon carbide nanotubes (SiCNTs) and nanowires, synthesized in various shapes and structures are often used as an alternate to CNTs. It has been established9–12 that SiCNTs are excellent sensors for detecting small molecules like NO, CO, CO2, HCN, and NO2. Phenol gets adsorbed on (8, 0) SiCNT preferentially through OH–Si interaction rather than π–π interaction between the phenol ring and the hexagon of SiCNT.13 The armchair nanotubes of germanium carbide (GeCNT), whose elements belong to the same group 14, have been found to be semiconducting in nature with a wide spectrum of band gaps.14 Recently, electron transport properties and adsorption sensitivities of zigzag GeCNT towards N2, CO, SO2, HCN, NH3, and H2CO molecules have also been theoretically studied.15,16In addition to the nanotubes of group 14, attention has been drawn to the nanotubes formed by group 13 and 15, namely, boron nitride nanotubes (BNNTs). The BNNTs are found to be less toxic compared to CNTs, and they also exhibit electronic properties independent of the diameter and chirality. It has been theoretically17 revealed that at the ambient conditions, boron-rich BNNT can capture CO2 initiated through the barrier-less physisorption process. A DFT study18 shows that the pristine and cobalt doped BNNTs interact with CO2 more strongly compared to CNTs, and hence can be used as a better absorber. Xiao et al.19 have performed ONIOM calculations to show that NO and NO2 can be used successfully to heal the N-vacancy in BNNT.
Gallium phosphide being isovalent with BN has been found to be useful for light emission devices in the visible range.20 GaP nanowires and nitrogen doped nanobelts have been prepared in recent years.21–25 Gallium phosphide nanotubes (GaPNTs) with a zinc blend structure have been synthesized and characterized by Wu et al.26 The structures of (10, 0) and (6, 6) GaPNTs are optimized from DFT calculations by Mirzaei and Mirzaei.27 These authors have also calculated NMR properties of (6, 0) and (4, 4) GaPNTs, including isotropic and anisotropic chemical shielding parameters.28 Effects of tubular diameters on the electronic and structural properties of GaPNTs have been investigated theoretically.29 All electron based DFT calculations with GGA (PBE) exchange correlation functional are carried out30 to optimize the geometries of alkali and transition metal doped single walled GaPNT (10, 0). Srivastava et al.31 have studied the stabilities and electronic properties of zigzag GaPNTs by ab initio based DFT calculations. These authors have also used a two-probe model for (4, 0) GaPNT to explore its transport properties. In this article, we report the adsorption sensitivities of several toxic molecules such as CO, HCN, SO2, NH3, and H2CO on (n, 0) GaPNTs (n = 5–7) using DFT based ONIOM calculations. Adsorption energy, Gibbs free energy change, and charge transfer from nanotube to the adsorbate molecule are calculated in each interaction.
2. Computational details
Three semiconducting zigzag (5, 0), (6, 0), and (7, 0) GaPNTs are considered in the present study. Preliminary calculations have been carried out on perfect GaPNTs with PBE32 functional in DFT using SIESTA-3.1 code33 to get their appropriate input geometries for the actual QM/MM calculations of the nanotube–adsorbate complex. Adsorptions of small molecules towards (n, 0) GaPNTs are studied here by adopting a two-layered ONIOM approach.34,35 Although the ONIOM method is not efficient to handle truly delocalized properties like band structure, several successful studies have been performed to obtain accurate results for large complex molecular systems in different chemical problems, including thermochemistry, homogeneous catalysis, stereoselectivity in organic synthesis, structure and enzymatic reaction mechanism.36 Geometries of nanotube–adsorbate complexes are optimized for various orientations of the axis of the adsorbate molecule. We have explored different adsorption sites such as (i) top of the P/Ga atom, (ii) top of the center of the Ga–P bond, and (iii) top of the center of the Ga–P hexagon. In the two-layer ONIOM calculation, the DFT method of the hybrid density functional, B3LYP37–39 with 6-31G(d) basis set for all atoms is employed for the high layer, while for the low layer a molecular mechanics method with Universal Force Field (UFF)40 is used. It is difficult to give a preference to molecular mechanics over the semiempirical method like PM6 for the low layer in the ONIOM calculations. The calculations with PM6 are computationally much more demanding than the UFF based calculations. Due to the problem of the SCF convergence in the PM6 method, the ONIOM optimization and frequency analysis could not be successfully carried out for some of the GaPNT–adsorbate complexes. Again in view of the cost-efficiency, the lower level description by UFF is found to be attractive. The ONIOM methodology as implemented in Gaussian 03 (ref. 41) uses such a QM/MM approach. The energy of a given system in a two-layer ONIOM approach is given by| E [ONIOM] = Ereal [MM] + Emodel [QM] − Emodel [MM], |
| Ea = E [ONIOM]GaPNT–adsorbate − E [B3LYP/6-31G(d)]adsorbate − E [ONIOM]GaPNT |
A negative value of Ea signifies a favorable adsorption. Adsorption energies are computed without using counterpoise (CP) corrections to the basis set superposition error (BSSE) as these cannot be done in the ONIOM approach. Moreover, it was previously shown that CP corrections do not improve energies and the uncorrected results are in better agreement with the experimental data.46,47 In order to check the thermodynamic spontaneity of the process, Gibbs free energy changes (ΔGa) due to molecular adsorption are calculated from the following expression:
| ΔGa = G(GaPNT–adsorbate) − G(GaPNT) − G(adsorbate) |
3. Results and discussion
Fig. 1 shows the most stable structure of the perfect (6, 0) GaP nanotube optimized at the ONIOM (B3LYP/6-31G(d):UFF) level. Three typical Ga–P bonds (labeled as ‘a’, ‘b’, and ‘c’) within this high layer are not exactly the same. The average Ga–P bond length is very close to the previously reported value of 2.28 Å by Mirzaei and Mirzaei.27 In accordance with the earlier calculations31 on (n, 0) GaPNTs (3 ≤ n ≤ 16) at the PBE/DZP level of theory, the present QM/MM study also shows that after optimization the more electronegative P atoms move outward radially, while the more electropositive Ga atoms shift inward of the curved hexagonal layer. A different hybridization of Ga and P results in the buckling of the tube surface and the degree of buckling reduces with the chirality. Each of CO, HCN, SO2, NH3, and H2CO is allowed to be adsorbed on the pyrene-like ring of the nanotube. The relaxed high-layer structures optimized at the B3LYP/6-31G(d) level of theory for the adsorption of all five molecules on (5, 0) GaPNT are shown in Fig. 2. | ||
| Fig. 1 Highlighted atoms of the representative perfect GaP nanotube used in the high layer of the ONIOM calculation. The pyrene-like structure is used for the adsorption of a single adsorbate molecule. (Color codes: Ga – pink, P – yellow.) | ||
 | ||
| Fig. 2 Optimized high-layer structures for the adsorption of single molecule on the (5, 0) GaPNT surface. | ||
In the CO adsorbed complex, carbon binds relatively strongly with Ga keeping the oxygen atom away from the nanotube. The CO molecule is positioned on top of the Ga atom of the hexagonal layer. Ga–C–O is almost linear with an average bond angle of about 177° and the neighboring Ga–P bonds relax marginally. In the most stable SO2 adsorbed complex of GaPNT, the sulphur atom is strongly bonded to the central P atom of the pyrene ring, while the two O atoms are attached to the two adjacent Ga atoms. A slight difference in the bond distances of the two Ga–O bonds may be due to the choice of the model in the high layer. The P–S and Ga–O interaction distances increase slowly with the increase in the diameter of the nanotube. The adsorption of SO2 is expected to be stronger for (5, 0) GaPNT. Due to strong adsorption, the neighboring Ga–P bonds elongate by 0.14–0.18 Å for all the GaPNTs studied here.
The adsorption of HCN on GaPNT is reasonably strong. The N atom of HCN is oriented towards the central Ga of the pyrene ring. In the HCN adsorbed complex, Ga–N–C–H remains almost collinear for all three chiralities and the neighboring Ga–P bonds relax by about 0.03–0.04 Å due to this adsorption. The ammonia molecule gets adsorbed to the GaP nanotube very strongly. The N atom is bonded directly to the central Ga atom with an average Ga–N distance of 2.18 Å. Three symmetrical H atoms are oriented above the pyrene ring of the nanotube in a staggered conformation with respect to three Ga–P bonds, which make 94–98° with Ga–N.
When H2CO is adsorbed on the GaP nanotube, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is oriented parallel to the Ga–P bond making a four-membered ring in the most stable configuration and the reaction is similar to a [2 + 2] cycloaddition. The C atom of the formaldehyde molecule is bonded to the P atom in the middle of the pyrene ring, while the O atom is attached to Ga. The C–O bond (labeled as ‘b’) in the adsorbed nanotube is elongated by about 0.16 Å, while the H–C–H bond angle is reduced to 109° due to the change in the hybridization. The elongation of the surrounding Ga–P bonds due to adsorption of H2CO is 0.07–0.12 Å. For all three chiralities, a second most stable configuration of H2CO-adsorbed nanotube has been predicted, in which C and O atoms of H2CO are bonded to Ga and P atoms of the nanotube, respectively, making a similar type of [2 + 2] cycloaddition. The energy of this second complex is calculated to be 83–92 kJ mol−1 higher than the most stable one.
O is oriented parallel to the Ga–P bond making a four-membered ring in the most stable configuration and the reaction is similar to a [2 + 2] cycloaddition. The C atom of the formaldehyde molecule is bonded to the P atom in the middle of the pyrene ring, while the O atom is attached to Ga. The C–O bond (labeled as ‘b’) in the adsorbed nanotube is elongated by about 0.16 Å, while the H–C–H bond angle is reduced to 109° due to the change in the hybridization. The elongation of the surrounding Ga–P bonds due to adsorption of H2CO is 0.07–0.12 Å. For all three chiralities, a second most stable configuration of H2CO-adsorbed nanotube has been predicted, in which C and O atoms of H2CO are bonded to Ga and P atoms of the nanotube, respectively, making a similar type of [2 + 2] cycloaddition. The energy of this second complex is calculated to be 83–92 kJ mol−1 higher than the most stable one.
Table 2 displays the computed adsorption energies (Ea) and Gibbs free energy changes (ΔGa) for the adsorption of one adsorbate molecule on (n, 0) GaPNTs (n = 5–7) at the ONIOM (B3LYP/6-31G(d):UFF) level of theory. Looking at the magnitude of the adsorption energies, it is predicted that the strongest adsorption occurs with the NH3 molecule. The GaP nanotube of smaller chirality is found to be susceptible to stronger adsorption of these small molecules. However, the calculated ΔGa values at T = 298.15 K for the adsorption of SO2 and NH3 are largely negative, while for the adsorption of CO and H2CO on (n, 0) GaPNTs, these are positive, implying that these adsorption processes are not thermodynamically favorable. In case of the adsorption of HCN, ΔGa for (5, 0) GaPNT is only −2.8 kJ mol−1, while for (6, 0) and (7, 0) nanotubes these are 1.3 and 6.8 kJ mol−1, respectively. We searched for any possible transition structure during the adsorption of all five small molecules, but found none. Therefore, it is expected that none of the adsorption processes studied here require crossing a barrier. The negative value of ΔGa signifies the exergonic adsorption process, and hence is a measure of thermodynamic spontaneity. Electronic interaction energies are always negative, but ΔH − TΔS determines the course of the reaction. Thus, the positive free energy changes in case of CO and H2CO indicate that the adsorptions may not occur under the conditions for which the calculations were made. However, the actual experimental conditions will dictate the spontaneity of the adsorption processes.
| Adsorbate molecule | Bondsa | Bond length (Å) | ||
|---|---|---|---|---|
| (5, 0) GaPNT | (6, 0) GaPNT | (7, 0) GaPNT | ||
| a Fig. 2.b Values in parentheses are results obtained using wB97XD functional. | ||||
| CO | a | 2.719 | 2.807 | 2.905 |
| (2.465) | (2.575) | (2.675) | ||
| b | 1.134 | 1.134 | 1.135 | |
| (1.129) | (1.130) | (1.130) | ||
| SO2 | a | 2.238 | 2.242 | 2.244 |
| (2.186) | (2.186) | (2.185) | ||
| b | 1.588 | 1.585 | 1.582 | |
| (1.571) | (1.572) | (1.571) | ||
| c | 2.007 | 2.008 | 2.016 | |
| (2.004) | (2.002) | (2.017) | ||
| d | 1.569 | 1.568 | 1.567 | |
| (1.560) | (1.559) | (1.557) | ||
| e | 2.035 | 2.035 | 2.044 | |
| (2.005) | (2.008) | (2.017) | ||
| HCN | a | 2.200 | 2.215 | 2.245 |
| (2.162) | (2.167) | (2.187) | ||
| NH3 | a | 2.150 | 2.157 | 2.171 |
| (2.120) | (2.123) | (2.136) | ||
| H2CO | a | 1.926 | 1.928 | 1.935 |
| (1.906) | (1.904) | (1.910) | ||
| b | 1.370 | 1.369 | 1.364 | |
| (1.373) | (1.373) | (1.370) | ||
| c | 1.980 | 1.983 | 1.991 | |
| (1.935) | (1.933) | (1.936) | ||
| Adsorbate molecule | (5, 0) GaPNT | (6, 0) GaPNT | (7, 0) GaPNT | |||
|---|---|---|---|---|---|---|
| Ea | ΔGa | Ea | ΔGa | Ea | ΔGa | |
| CO | −28.5 | 6.2 | −25.6 | 8.8 | −21.9 | 12.1 |
| SO2 | −81.4 | −25.0 | −77.2 | −21.0 | −67.3 | −12.3 |
| HCN | −42.3 | −2.8 | −38.2 | 1.3 | −32.9 | 6.8 |
| NH3 | −98.6 | −62.6 | −97.2 | −57.8 | −87.9 | −48.2 |
| H2CO | −14.9 | 33.3 | −11.9 | 36.6 | −4.7 | 42.8 |
We have extended our ONIOM calculations on the adsorptions of all five molecules on (5, 0) GaPNT using TZVP basis sets to explore the improvement of their adsorption energies, if any. A full geometry optimization calculation has been carried out at the ONIOM(B3LYP/TZVP:UFF) level. The comparative results are shown in ESI Fig. S1,† which indicate that B3LYP/TZVP calculations overestimate the interaction energy by 22–28 kJ mol−1.
Fig. 3 displays the density of states (DOS) spectra of the bare and adsorbed (5, 0) GaPNT, while the remaining two sets of DOS spectra for (6, 0) and (7, 0) nanotubes are shown in ESI Fig. S2 and S3.† Each DOS spectrum is convoluted with a Gaussian FWHM of 0.3 eV and the zero energy is set to HOMO. In general, the computed HOMO–LUMO gap (Eg) of (n, 0) GaPNT gradually increases with the diameter of the tube (Table 3). This is in accordance with the results obtained by Srivastava et al.31 based on band structure analysis using the k-point sampling method. As a result of the adsorption of one adsorbate molecule, Eg is increased by 0.06–0.24 eV. Only in case of the adsorption of SO2 on (7, 0) GaPNT, the computed Eg is reduced by 0.13 eV. The HOMO peak looks similar for all single molecule adsorptions except SO2. The band splitting in the upper part of the valence band and in the lower part of the conduction band are noticeable due to the stronger adsorption of the sulphur dioxide molecule on (5, 0) GaPNT. The density around HOMO is also changed for NH3 indicating its larger influence on the electronic structure of the nanotube. However, the signature of the DOS spectra varies with the diameter of the tube, but in each case the overall spectra of SO2 and NH3 adsorbed complexes are different compared to those of the corresponding bare nanotube.
 | ||
| Fig. 3 DOS spectra of the adsorbed (5, 0) GaPNT complexes. | ||
| Adsorbate molecule | (5, 0) GaPNT | (6, 0) GaPNT | (7, 0) GaPNT | |||
|---|---|---|---|---|---|---|
| Eg | QT | Eg | QT | Eg | QT | |
| CO | 2.14 | 0.14 | 2.20 | 0.13 | 2.57 | 0.12 |
| SO2 | 2.15 | −0.31 | 2.20 | −0.31 | 2.38 | −0.31 |
| HCN | 2.23 | 0.17 | 2.28 | 0.17 | 2.68 | 0.16 |
| NH3 | 2.21 | 0.24 | 2.27 | 0.24 | 2.69 | 0.25 |
| H2CO | 2.32 | −0.34 | 2.39 | −0.34 | 2.54 | −0.34 |
| Pure nanotube | 2.08 | 2.15 | 2.51 | |||
The extent of charge transfer (QT) between GaPNT and the adsorbate molecule is calculated from the Mulliken analysis and the computed values for all three GaP nanotubes are given in Table 3. The negative value of QT implies that the electron is transferred from the nanotube to the adsorbate molecule. SO2 and H2CO molecules after being adsorbed withdraw about 0.31e and 0.34e charges, respectively from the GaP nanotube. For the adsorption of other three molecules, viz. CO, HCN, and NH3, the nanotube as a whole accumulates some amount of electronic charges. The amount of charge transfer does not vary much with the size of the tube.
When the dispersion interactions are included through the wB97XD functional, the important interaction bond distances of the adsorbed complexes shorten. This has been shown in Table 1 by comparing these bond distances from the ONIOM calculations using B3LYP and wB97XD functionals, respectively. ESI Tables S1 and S2† display other results of the calculations, which include such dispersion interactions. However, QT values computed using wB97XD functional do not change much. As depicted in Fig. 4, despite having an excellent matching of QT, the calculated adsorption energies using B3LYP hybrid functional are overestimated by as much as 41 kJ mol−1. This overestimation is reflected in the large reduction of Eg.45 The computed Eg values of different adsorbing species obtained by employing both B3LYP and wB97XD functionals are shown in Fig. 5. The relative values for different adsorbate molecules in two different functional are comparable.
 | ||
| Fig. 4 Comparison of (a) adsorption energy (Ea) and (b) Mulliken charge transfer (QT) computed with B3LYP and wB97XD functionals. | ||
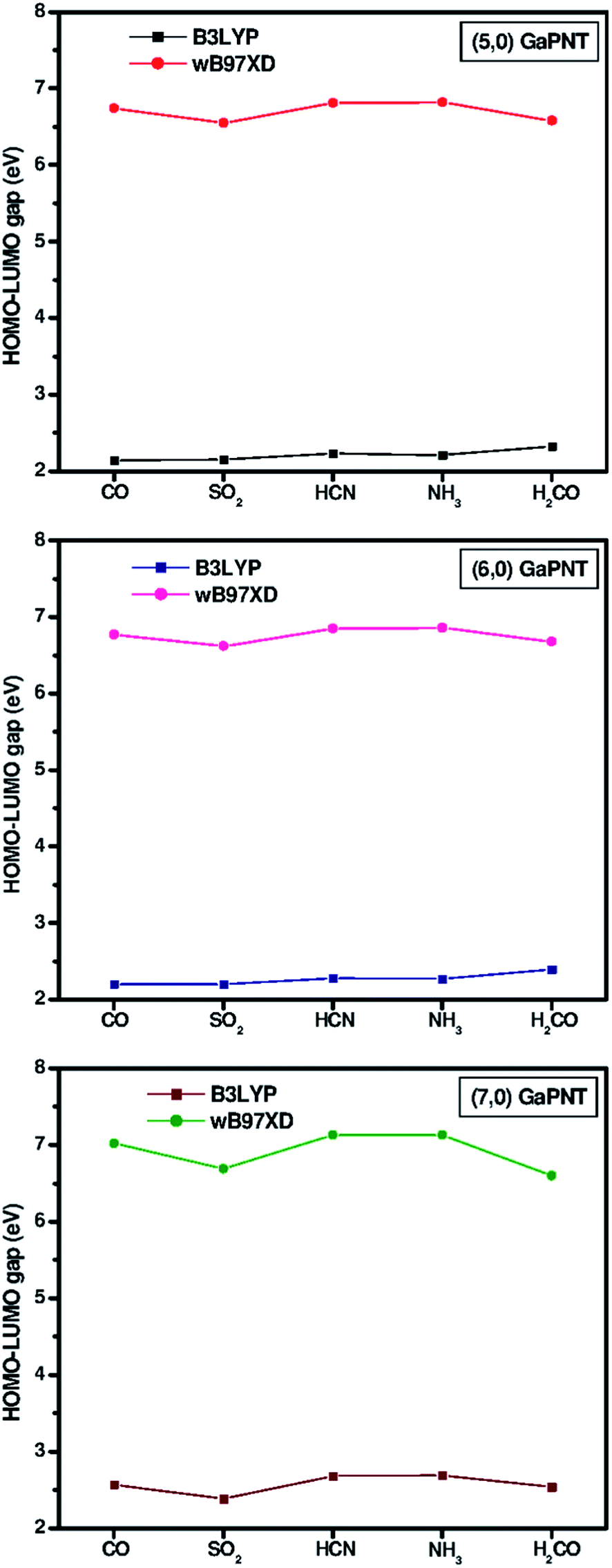 | ||
| Fig. 5 Comparison of the HOMO–LUMO gap (Eg) computed with B3LYP and wB97XD functional. | ||
4. Conclusion
Adsorptions of small molecules, viz. CO, SO2, HCN, NH3, and H2CO, on (n, 0) GaPNTs (n = 5–7) have been investigated by carrying out ONIOM calculations at the B3LYP/6-31G(d):UFF level. All the adsorption processes are found to be exothermic in nature and their energy profiles do not possess any energy barrier as no transition structure has been obtained. In general, bindings of NH3, SO2, and HCN towards GaP nanotubes are predicted to be stronger than either CO or H2CO molecules. The smaller nanotubes are more reactive to these adsorbate molecules. The changes in the DOS pattern of the adsorbed GaPNTs help in analyzing the extent of binding between the nanotube and the adsorbate molecule. The HOMO–LUMO gap of the GaP nanotube generally increases upon the adsorption of small molecule. The adsorption of SO2 on (7, 0) GaPNT is an exception for which the gap decreases by about 0.13 eV. Mulliken charge analyses predict that the adsorptions of SO2 and H2CO result in charge transfer from GaPNT to the adsorbate molecule. In the adsorption of other molecules, the charge is transferred in the opposite direction. The adsorption energies computed at the ONIOM(B3LYP/6-31G(d):UFF) level of theory are always overestimated compared to ONIOM(wB97XD/6-31G(d):UFF) calculations. The dispersive energy dominates for the adsorption of gas molecules on to the GaPNT surface. However, in the latter case, the HOMO–LUMO energy gaps are larger than those obtained with pure and hybrid density functional. Of all the GaPNTs studied here, the Eg of (5, 0) GaPNT increases by about 11.5% upon the adsorption of H2CO molecule.References
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus and P. Avouris, Carbon Nanotubes: Synthesis, Structure, Properties and Applications, Springer, Berlin, 2000 Search PubMed.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chapline, S. Peng, K. J. Cho and H. J. Dai, Science, 2000, 287, 622 CrossRef CAS.
- P. G. Collins, K. Bradley, M. Ishigami and A. Zettl, Science, 2000, 287, 1801 CrossRef CAS.
- A. Goldoni, R. Larciprete, L. Petaccia and S. Lizzit, J. Am. Chem. Soc., 2003, 125, 11329 CrossRef CAS PubMed.
- J. Zhao, A. Buldum, J. Han and J. P. Lu, Nanotechnology, 2002, 13, 195 CrossRef CAS.
- G. Guo, F. Wang, H. Sun and D. Zhang, Int. J. Quantum Chem., 2008, 108, 203 CrossRef CAS.
- H. Jiang, D. Zhang and R. Wang, Nanotechnology, 2009, 20, 145501 CrossRef PubMed.
- R. Q. Wu, M. Yang, Y. H. Lu, Y. P. Feng, Z. G. Huang and Q. Y. Wu, J. Phys. Chem. C, 2008, 112, 15985 CAS.
- G. H. Gao and H. S. Kang, J. Chem. Theory Comput., 2008, 4, 1690 CrossRef CAS.
- G. H. Gao, S. H. Pank and H. S. Kang, Chem. Phys., 2009, 355, 50 CrossRef CAS PubMed.
- J. X. Zhao and Y. H. Ding, J. Chem. Theory Comput., 2009, 5, 1099 CrossRef CAS.
- J. X. Zhao, B. Gao, Q. H. Cai, X. G. Wang and X. Z. Wang, Theor. Chem. Acc., 2011, 129, 85 CrossRef CAS.
- S. J. Rathi and A. K. Ray, Nanotechnology, 2008, 19, 335706 CrossRef PubMed.
- P. N. Samanta and K. K. Das, J. Phys. Chem. C, 2012, 117, 515 Search PubMed.
- P. N. Samanta and K. K. Das, Chem. Phys. Lett., 2013, 577, 107 CrossRef CAS PubMed.
- H. Choi, Y. C. Park, Y.-H. Kim and Y. S. Lee, J. Am. Chem. Soc., 2011, 133, 2084 CrossRef CAS PubMed.
- E. N. C. Paura, W. F. Da Cunha, J. B. L. Martins, G. M. E Silva, L. F. Roncaratti and R. Gargano, RSC Adv., 2014, 4, 28249 RSC.
- B. Xiao, X.-F. Yu and Y.-H. Ding, RSC Adv., 2014, 4, 22688 RSC.
- Y. H. Kim, Y. W. Jun, B. H. Jun, S. M. Lee and J. Cheon, J. Am. Chem. Soc., 2002, 124, 13656 CrossRef CAS PubMed.
- C. Tang, S. Fan, M. L. de la Chapelle, H. Dang and P. Li, Adv. Mater., 2000, 12, 1346 CrossRef CAS.
- S. Gao, Y. Xie, J. Lu, G. Du, W. He, D. Cui, B. Huang and M. Jiang, Inorg. Chem., 2002, 41, 1850 CrossRef CAS PubMed.
- W. S. Shi, Y. F. Zheng, N. Wang, C. S. Lee and S. T. Lee, Adv. Mater., 2001, 13, 591 CrossRef CAS.
- H. W. Seo, S. Y. Bae, J. Park, H. Yang, M. Kang and S. Kim, Chem. Commun., 2002, 2564 RSC.
- H. W. Seo, S. Y. Bae, J. Park, H. Yang, M. Kang and S. Kim, Appl. Phys. Lett., 2003, 82, 3752 CrossRef CAS PubMed.
- Q. Wu, Z. Hu, C. Liu, X. Wang, Y. Chen and Y. Lu, J. Phys. Chem. B, 2005, 109, 19719 CrossRef CAS PubMed.
- M. Mirzaei and M. Mirzaei, Phys. E, 2011, 43, 1343 CrossRef CAS PubMed.
- M. Mirzaei and M. Mirzaei, Monatsh. Chem., 2011, 142, 111 CrossRef CAS.
- M. Mirzaei, M. Yousefi and M. Meskinfam, Solid State Sci., 2012, 14, 801 CrossRef CAS PubMed.
- C. Kamal, A. Chakrabarti, A. Banerjee and S. K. Deb, Phys. E, 2013, 54, 273 CrossRef CAS PubMed.
- A. Srivastava, S. K. Jain and P. S. Khare, J. Mol. Model., 2014, 20, 2171 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef CAS.
- S. Daprich, I. Komáromi, K. S. Byun, K. Morokuma and M. J. Frisch, J. Mol. Struct.: THEOCHEM, 1999, 462, 1 CrossRef.
- T. Vreven, K. S. Byun, I. Komáromi, S. Daprich, J. A. Montgomery Jr, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815 CrossRef CAS.
- K. Morokuma, Bull. Korean Chem. Soc., 2003, 24, 797 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef.
- M. J. Frischet al., GAUSSIAN 03 revision E.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. Ricca and J. A. Drocco, Chem. Phys. Lett., 2002, 362, 217 CrossRef CAS.
- R. Wanbayor and V. Ruangpornvisuti, Carbon, 2008, 46, 12 CrossRef CAS PubMed.
- X. Lu, F. Tian, X. Xu, N. Wang and Q. Zhang, J. Am. Chem. Soc., 2003, 125, 10459 CrossRef CAS PubMed.
- S. Osuna, J. Morera, M. Cases, K. Morokuma and M. Sola, J. Phys. Chem. A, 2009, 113, 9721 CrossRef CAS PubMed.
- J. R. Alvarez-Idaboy and A. Galano, Theor. Chem. Acc., 2010, 126, 75 CrossRef CAS.
- L. M. Mentel and E. J. Baerends, J. Chem. Theory Comput., 2014, 10, 252 CrossRef CAS.
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio molecular orbital theory, Wiley, New York, 1986, pp. 226–261 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09706h |
| This journal is © The Royal Society of Chemistry 2014 |