
A facile fluorescence method for endonuclease detection using exonuclease III-aided signal amplification of a molecular beacon†
Chan Song ab,
Qi Zhangab,
Gui-Mei Hanab,
Yi-Chen Duab and
De-Ming Kong*ab
ab,
Qi Zhangab,
Gui-Mei Hanab,
Yi-Chen Duab and
De-Ming Kong*ab
aState Key Laboratory of Medicinal Chemical Biology, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: kongdem@nankai.edu.cn; Fax: +86-22-23502458; Tel: +86-22-23500938
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, China
First published on 10th October 2014
Abstract
An exonuclease III (Exo III)-aided signal amplified endonuclease detection strategy was developed by employing a hairpin DNA as the substrate for endonucleases. In the presence of endonucleases, the elaborately designed stem-loop substrate was cleaved into two parts. With the help of Exo III, a single-stranded target of a molecular beacon (MB) probe was released. Subsequently, the MB would hybridize with this target to form a double-stranded DNA, opening its hairpin structure and thereby resulting in the restoration of the fluorescence signal. Owing to the presence of a recessed 3′ terminus in the formed double-stranded DNA, Exo III-aided recyclable cleavage of MBs was achieved. Eventually, an amplified fluorescence signal was observed. As a proof of concept, the fluorescence sensor of endonuclease EcoRI was designed. Under the optimized conditions, the fluorescence intensity is linear with EcoRI activity over the wide range of 1 to 80 U mL−1, with a detection limit of 0.57 U mL−1. Besides, detection of BamHI activity with satisfactory results was achieved by simply changing the endonuclease recognition sequence in the unlabelled substrate, demonstrating that the design concept can be widely adapted to other restriction endonuclease activity analysis.
Introduction
Endonucleases are widely used enzymes in modern molecular biology as they play important roles in many biological processes, including DNA replication, recombination, repair, molecular cloning, genotyping and mapping.1 Among those various endonucleases, Type II restriction endonucleases, could recognize short, usually palindromic sequences of 4–8 bp and, in the presence of Mg2+, cleave the DNA within or in the proximity of the recognition sequence.2 Moreover, Type II restriction endonucleases are also powerful tools in the studies of DNA–protein interactions3 and medicinal chemistry.4 Importantly, these restriction endonucleases have been employed as versatile targets for antimicrobial and antiviral drug development.4a–d In addition, they play a significant role in protecting the host genome against foreign DNAs.4e,f Therefore, the development of novel methods for sensitively assaying activities of these enzymes is of great significance in the fields of biotechnology and drug discovery.Traditional analytical methods, including gel electrophoresis, chromatography, immune reaction and radioactive labeling etc., have been reported to detect endonuclease activities. Although sensitive, their complicated, time-consuming, laborious operation and the requirement for radioactive labelling greatly limit their widespread applications.5 Up to now, many novel detection strategies are springing up to circumvent above drawbacks.6 For example, colorimetric approaches based on gold nanoparticles are widely used in detecting endonucleases.6a–c Despite that the sensitivity of these methods is acceptable, their handling procedures are still complicated and time-consuming, hindering their practical applications. Thus, it is still urgent to develop convenient sensing platforms for sensitive detection of those enzyme activities. Recently, many oligonucleotide-based optical sensing platforms for assaying endonuclease activity have been reported.6d–f For example, Dong and Ma et al. used G-quadruplex DNA as the fluorescence signal reporters for the detection of endonucleases.6e,f Moreover, the dye-labelled DNAs were widely utilized in the design of the nanomaterial-based sensing platforms.6g–i The activity of endonucleases could be successfully detected by taking advantages of the specific recognition ability of endonucleases and the typical intrinsic optical, electronic and catalytic characteristics of nanomaterials. As fluorescent assay methods have the inherent merits of operation convenience, rapid kinetics and ease of automation,7 the fluorescent assay methods, especially the fluorescent amplification methods, are becoming increasingly popular.6j–l
Recently, the enzyme-assisted signal amplification strategies involving DNA enzymes, such as polymerase, nicking endonucleases and exonuclease III (Exo III), have been successfully used to improve the detection sensitivity of nucleic acids,8 protein,9 small molecules10 and metal ions.11 As the polymerase-based amplification methods easily suffer from cross-contamination in the amplification process, a few of complex operation processes are required to overcome this issue, thus resulting in high cost or false-positive results.12 In the nicking endonuclease methods, a target DNA with a specific enzyme recognition sequence is required. Exonuclease III (Exo III) is a kind of exonuclease that can catalyze the stepwise removal of mononucleotides from 3′-hydroxyl termini of duplex DNAs.13 Different from nicking endonucleases, Exo III does not require any specific recognition sequences due to its sequence-independent enzyme activity, which makes the Exo III-catalyzed amplification platform simple, fast and cost-effective. Therefore, the Exo III-catalyzed amplification strategies are attracting more and more interest.14
Herein, we constructed a molecular beacon-based fluorescence assay method for simple and rapid detection of endonuclease activity using Exo III as a signal amplifier. As an example, the activity of endonuclease EcoRI has been quantitatively analyzed within a short assay time in a single tube. The detection limit was 0.57 U mL−1 with a wide linear range from 1 to 80 U mL−1. The selectivity of the designed sensing system was also tested with satisfactory results. Furthermore, to demonstrate the generality of the sensor design strategy, the activity of endonuclease BamHI was successfully studied by simply changing the recognition sequence in an unlabelled DNA oligonucleotide. These results demonstrate the assay protocol presented here is simple, rapid, effective and easy to carry out.
Experimental section
Chemical and reagents
All oligonucleotides were purchased from Sangon Biotech. Co. Ltd. (Shanghai, China) and their sequences were listed in Table S1 (ESI†). The concentrations of the oligonucleotides were represented as single-stranded concentrations. Single-stranded concentrations were determined by measuring the UV absorbance at 260 nm. Exo III, restriction endonucleases (EcoRI, BamHI and HindIII), E. coli DNA ligase, phi29 DNA polymerase and T4 polynucleotide kinase (T4 PNK) were purchased from New England Biolabs (Beijing, China). Other chemicals were of analytical grade and used without further purification.Apparatus and measurements
All fluorescence measurements were carried out on a SHIMADZU RF-5301PC spectrofluorimeter with 1 cm-path-length micro quartz cell (40 μL, Starna Brand, England). The emission spectra were collected from 490 nm to 610 nm with an excitation wavelength of 480 nm at room temperature. The fluorescence intensity at 516 nm is used for quantitative analysis. The excitation and emission slit widths were both set at 5 nm.Assay of restriction endonuclease activity
Taking EcoRI activity assay as an example, a mixture of 100 nM Hairpin substrate 1 (Table S1, ESI,† Hairpin substrate 2 for BamHI activity analysis) and 200 nM MB (Table S1, ESI†) was prepared in 25 mM Tris–HCl buffer (pH 8.0) containing 100 mM NaCl and 5 mM MgCl2. Upon vortexing, the solution was heated to 95 °C for 5 min, cooled slowly to 25 °C and incubated at 25 °C for 30 min. Then, Exo III (2 U mL−1, final concentration) and different amounts of EcoRI were added to a final volume of 100 μL. After the solutions were incubated at 37 °C for 60 min, the fluorescence signal was recorded.Results and discussion
Principle of amplified endonuclease activity detection using Exo III and molecular beacon
Using endonuclease EcoRI as a model system, the working principle of Exo III-aided signal amplified endonuclease activity analysis is described in Scheme 1. Exo III, whose activity is sequence-independent, can catalyse the stepwise removal of mononucleotides from 3′-hydroxyl termini of duplex DNAs.13 Due to the limitation of its activity on single-stranded DNA, the enzyme's preferred substrates are 3′ blunt or recessed termini of duplex DNAs.13a,15 As can be seen, the hairpin structures of Hairpin substrate 1 and MB both have 3′ protruding terminus that could resist the digestion of Exo III. Hairpin substrate 1 has a long stem, which contains the recognition sequence 5′-GAATCC-3′ for EcoRI (the blue part, Scheme 1 and Table S1 in ESI†). Thus, in the presence of EcoRI, the cleavage reaction is initiated and Hairpin substrate 1 is cleaved into two parts (part A and part B, Scheme 1). In part B, only a 3-bp stem remained. Such a hairpin structure is unstable at the reaction temperature of 37 °C, and part B might exist as single-stranded structure in the ultimate. Part A is a 16-bp hybrid duplex with a recessed 3′ terminus. Exo III then can catalyse the stepwise removal of mononucleotides from this terminus of part A, releasing a single-stranded target of the probe MB. We named this single-stranded target as triggered target because it could trigger the digestion of MB. Subsequently, the hybridization between MB and the triggered target forms a double-stranded duplex structure with a recessed 3′ terminus, leading, in turn, to the digestion of MB by Exo III. Accompanied by the digestion of MB, the fluorophore (FAM) is completely separated from the quencher (DABCYL), resulting the recovery of the quenched fluorescence. Because the 3′ terminus of the triggered target is protruding, it cannot be digested by Exo III. As a result, the released target then hybridizes with another MB to perform recycling. Eventually, each EcoRI could remarkably lead to the digestion of many MB, affording a dramatically amplified fluorescence signal. However, in the absence of EcoRI, Hairpin substrate 1 cannot be cleaved and kept its own original hairpin shape with a long double-stranded stem. As the active binding sites on MB (red section, Scheme 1) are all protected in double-stranded form in the Hairpin substrate 1, there is a good stability between Hairpin substrate 1 and MB. Therefore, without the help of EcoRI, the direct interaction between Hairpin substrate 1 and MB is impossible, thus ensuring the low background of the sensing system. | ||
| Scheme 1 Working principle of EcoRI activity analysis based on Exo III-aided signal amplification strategy. | ||
To confirm the feasibility of the sensing system, the results of experiments performed under different conditions were compared. As shown in Fig. 1, in the absence of EcoRI, the fluorescence intensity was weak (curve a) because of the lack of the triggered target. As a contrary, the fluorescence intensity increased greatly with the introduction of EcoRI (curve c). In order to confirm that the fluorescence enhancement is really related with the restriction endonuclease activity of EcoRI, a control experiment was conducted by using heat-inactivated EcoRI instead of active EcoRI. As expected, no signal response to inactivated EcoRI was observed (curve b), the signal level is comparable to that of the blank sample (curve a). These results indicated that the increasing of fluorescence emission in EcoRI sensing system can really reflect the activity of this enzyme.
 | ||
| Fig. 1 Fluorescence emission spectra at different experimental conditions: (a) in the absence of EcoRI; (b) incubated with heat-inactivated EcoRI; (c) incubated with active EcoRI. [Hairpin substrate 1] = 100 nM, [MB] = 200 nM, [EcoRI] = 100 U mL−1, [Exo III] = 2.0 U mL−1. | ||
Optimization of detection conditions
To obtain the best analytical performance, the effects of experimental conditions were investigated. As Exo III plays an extremely important role in catalyzing the stepwise removal of mononucleotides from 3′-hydroxyl termini of duplex DNAs to generate the triggered target and construct a signal amplification platform, the influence of Exo III concentration was tested as shown in Fig. 2A. It can be seen that the fluorescence signal increased gradually with the concentration of Exo III. Although the fluorescence signal still followed an increasing trend with further increase of Exo III concentration, the highest signal-to-noise ratio F/F0 (F0 and F are the fluorescence intensities in the absence and presence of EcoRI, respectively) value was observed at 2 U mL−1 of Exo III (Fig. S1, ESI†). As a result, 2 U mL−1 of Exo III was used for the following investigations. | ||
| Fig. 2 Fluorescence intensity with different amounts of Exo III (A) and Hairpin substrate 1 (B). F0 and F are the fluorescence intensities in the absence and presence of 100 U mL−1 EcoRI, respectively. | ||
In this proposed assay method, the detection of EcoRI activity is on the basis of the cleavage of Hairpin substrate 1 by EcoRI and subsequent hybridization of the released triggered target with MB, the amounts of Hairpin substrate 1 and MB are important factors for the excellent sensing performance. As shown in Fig. 2B, the fluorescence signal increased with the Hairpin substrate 1 amount in the range of 25–100 nM and then reached a plateau in the presence of EcoRI. Due to the highest F/F0 value (Fig. S2, ESI†), 100 nM of Hairpin substrate 1 was used in subsequent experiments. On the other hand, it was observed that the fluorescence intensity was significantly enhanced as MB concentration increased in the sensing systems with EcoRI addition (Fig. 3A). Unfortunately, increasing MB dosage also resulted in the increase of background fluorescence signal (Fig. 3A). Thus, we chose 200 nM of MB in the following experiments to get the highest the signal-to-noise ratio (Fig. S3, ESI†).
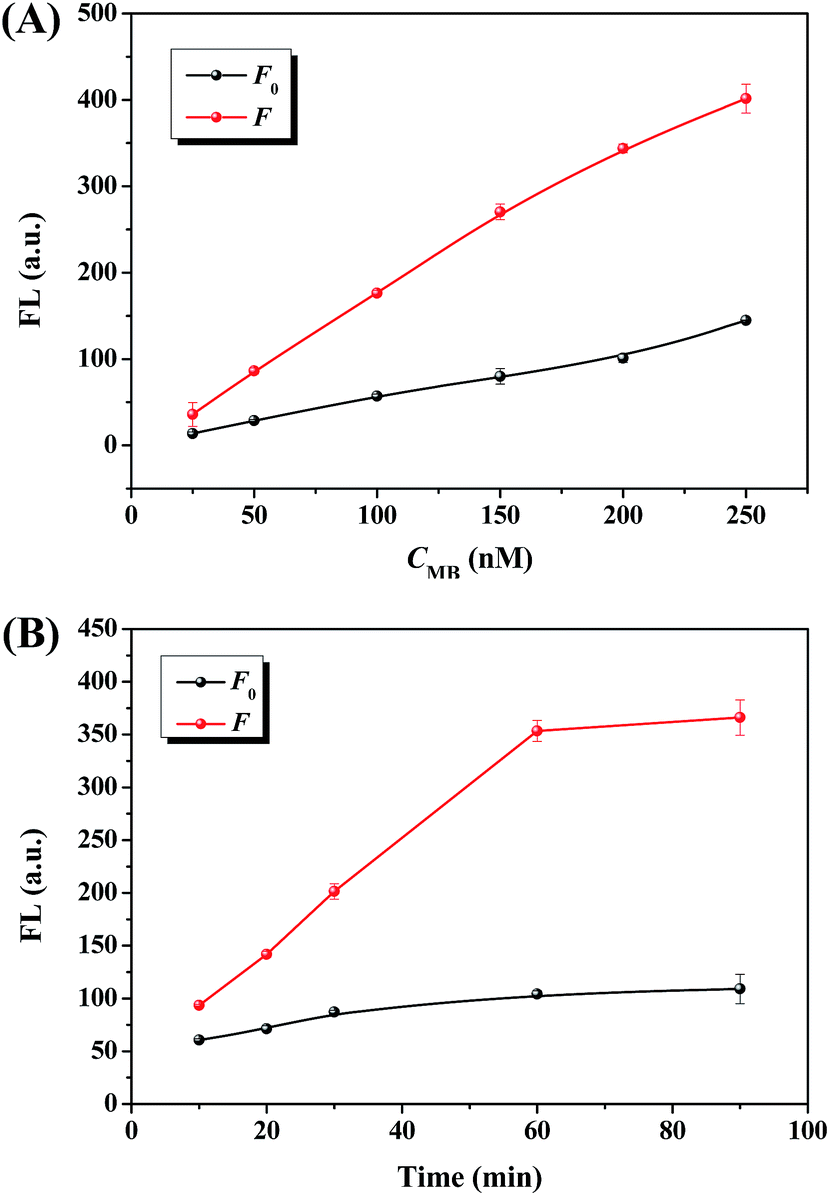 | ||
| Fig. 3 Fluorescent intensity with different amounts of MB (A) and versus incubation time at 37 °C (B). F0 and F are the fluorescence intensities in the absence and presence of 100 U mL−1 EcoRI, respectively. | ||
In order to obtain optimized assay time, the kinetic behaviour of the sensing platform was studied by monitoring the change of fluorescence intensity with time in the absence and presence of EcoRI, respectively. With the increase of incubation time, the fluorescence response to EcoRI increased quickly, and then leveled off to a plateau in 60 min (Fig. 3B). Nevertheless, in the absence of EcoRI, the fluorescence signal of the blank sample also showed a slight enhancement with time (Fig. 3B). As demonstrated in Fig. S4 (ESI†), the maximum F/F0 was observed in 60 min. So the optimized incubation time was 60 min.
As the specificity of enzymes can be affected by temperature, the effect of temperature on the sensing performance of this detection method was investigated as shown in Fig. S5 (ESI†). The fluorescence intensity gave the maximum enhancement relative to the blank control when the sample was cultured at 37 °C, indicating that Exo III and EcoRI exhibit the highest activity at this temperature. In addition, the maximum F/F0 was observed at this temperature, too. So this proposed activity assay method to the endonuclease was incubated at 37 °C.
Sensitivity and selectivity of the proposed EcoRI-sensing system
Under the optimized conditions, we measured the fluorescence response to different concentrations of EcoRI from 0 to 500 U mL−1. Obviously, fluorescence intensity at 516 nm increased as increasing the concentration of EcoRI (Fig. 4A), indicating that more and more fluorescent MB fragments were produced. As shown in Fig. 4B, a perfect linear relationship (R2 = 0.991) between the fluorescence intensity and EcoRI concentration was observed over the range of 1–80 U mL−1. From the slope of the fitting line of the data in the linear response region (k) and the standard deviation of blank samples measurement (σ), the detection limit of this EcoRI-sensing method was calculated to be 0.57 U mL−1 (3σ/k), which is much lower than that commonly used detection systems such as enzyme-linked immunosorbent assay (ELISA), high performance liquid chromatography (HPLC) and fluorescence resonance energy transfer (FRET) method.5,16 These results demonstrated high sensitivity and good linearity for EcoRI activity quantitation using this method. In addition, compared with traditional analytical methods, our method has the advantages of simplicity, time-saving and the ease of use.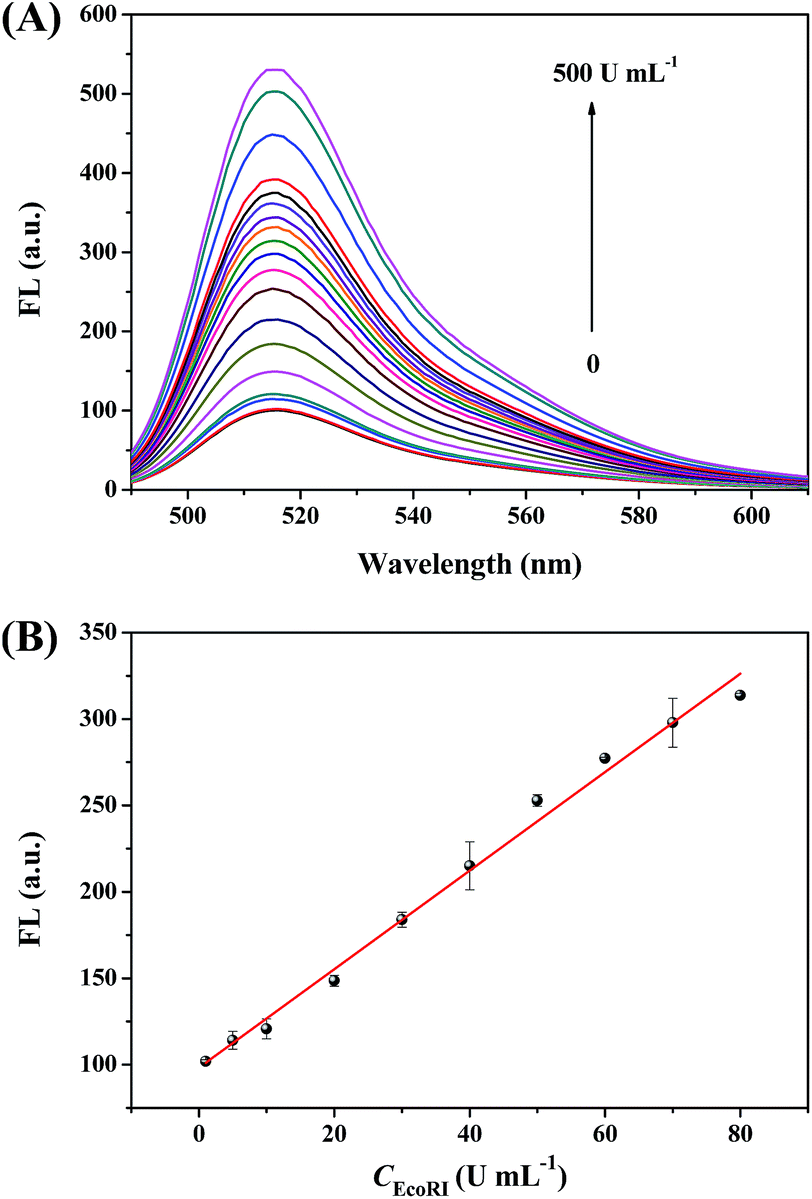 | ||
| Fig. 4 (A) Fluorescence emission spectra in the presence of different concentrations of EcoRI from 0 to 500 U mL−1. The activities of EcoRI are (arrow direction): 0, 1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120, 150, 200, 300 and 500 U mL−1. (B) The linear relationship between the fluorescence intensity and EcoRI concentration in the range of 1–80 U mL−1. [Hairpin substrate 1] = 100 nM, [MB] = 200 nM, [Exo III] = 2 U mL−1. All experiments were performed in triplicate. | ||
As the selectivity is one of the vital factors to estimate the performance of a sensing system, other enzymes, such as non-targeted endonucleases (including BamHI and HindIII), E. coli DNA ligase, phi29 DNA polymerase and polynucleotide kinase T4 PNK were chosen to evaluate the selectivity of the present approach (Fig. 5). As expected, remarkable fluorescence enhancement was obtained with the addition of EcoRI. However, the enhanced intensity for the tested enzymes was 0.9–3.9% of that for EcoRI at the same concentration. These results indicated that this Exo III-aided amplified sensing platform exhibited a good selectivity, which could attribute to the highly specific sequence recognition of EcoRI.
 | ||
| Fig. 5 Selectivity of the proposed EcoRI-sensing system. The concentrations of EcoRI, BamHI, HindIII, E. coli DNA ligase, phi29 DNA polymerase and T4 PNK are 100 U mL−1. [Hairpin substrate 1] = 100 nM, [MB] = 200 nM, [Exo III] = 2 U mL−1. | ||
Generality of the proposed sensing system
The proposed sensor design strategy can be easily extended to the assay of other restriction endonucleases. To verify its generality, a BamHI activity-sensing platform was designed, in which the unlabelled Hairpin substrate 1 was replaced by another unlabelled oligonucleotide (Hairpin substrate 2), and fluorescent labelled MB needed no change at all. Hairpin substrate 2 for BamHI activity assay was designed by simply replacing the recognition sequence of EcoRI (5′-GAATTC-3′, blue part in Table S1, ESI†) in the Hairpin substrate 1 by that of BamHI (5′-GGATTC-3′, pink part in Table S1, ESI†). As shown in Fig. 6, significant fluorescent enhancement was clearly observed by the introduction of BamHI. A good linear correlation (R2 = 0.994) was found between fluorescence intensity and BamHI concentration over the range from 1 to 100 U mL−1, with a detection limit of 0.60 U mL−1 (3σ/k). These results demonstrated that the proposed sensor design strategy has the potential to be a general method for the detection of a wide spectrum of restriction endonucleases by simply changing the enzyme recognition sequence in the unlabelled hairpin substrate. | ||
| Fig. 6 (A) Fluorescence emission spectra in the presence of different concentrations of BamHI. The activities of BamHI are (arrow direction): 0, 1, 5, 10, 20, 40, 60, 80 and 100 U mL−1. (B) The linear relation between the fluorescence intensity and BamHI concentration. [Hairpin substrate 2] = 100 nM, [MB] = 200 nM, [Exo III] = 2 U mL−1. All experiments were performed in triplicate. | ||
Conclusions
In summary, we have developed a novel molecular beacon-based sensing method for simple and rapid detection of the activity of restriction endonuclease via Exo III-aided signal amplification. Firstly, the protocol is easy to carry out in a single tube by simple mixing the enzymes and oligonucleotides, and the detection of target can be achieved within a short assay time. Secondly, by taking advantage of the cleavage function of restriction endonucleases on special sequences, the proposed strategy showed high selectivity. Thirdly, Exo III-aided signal amplification can provide the assay with high sensitivity. Finally, it was worthwhile to note that this new methodology can be easily extended to measure the activity of other restriction endonucleases by simply changing the recognition sequence of unlabelled substrate DNA. Overall, the proposed sensing system demonstrates great potential for further application in biological research.Acknowledgements
This work was support by the National Basic Research Program of China (no. 2011CB707703), the Natural Science Foundation of China (no. 21175072, 21322507), the National Natural Science Foundation of Tianjin (no. 12JCYBJC13300).Notes and references
- (a) A. B. Houtsmuller, S. Rademakers, A. L. Nigg, D. Hoogstraten, J. H. J. Hoeijmakers and W. Vermeulen, Science, 1999, 284, 958 CrossRef CAS; (b) H. J. Liu, J. H. Chen, M. H. Liao, M. Y. Lin and G. N. Chang, J. Virol. Methods, 1999, 81, 83 CrossRef CAS; (c) N. D. F. Grindley, K. L. Whiteson and P. A. Rice, Annu. Rev. Biochem., 2006, 75, 567 CrossRef CAS PubMed; (d) A. Z. Al-Minawi, Y.-F. Lee, D. Hakansson, F. Johansson, C. Lundin, N. Saleh-Gohari, N. Schultz, D. Jenssen, H. E. Bryant, M. Meuth, J. M. Hinz and T. Helleday, Nucleic Acids Res., 2009, 37, 6400 CrossRef CAS PubMed; (e) W. Choi and R. M. Harshey, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10014 CrossRef CAS PubMed.
- A. Pingoud and A. Jeltsch, Nucleic Acids Res., 2001, 29, 3705 CrossRef CAS PubMed.
- C. R. Robinson and S. G. Sligar, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2186 CrossRef CAS.
- (a) D. R. Lesser, M. R. Kurpiewski and L. Jen-Jacobson, Science, 1990, 250, 776 CAS; (b) P. A. Johnston, Drug Discovery Today, 2002, 7, 353 CrossRef CAS; (c) D. L. Boger, J. Desharnais and K. Capps, Angew. Chem., Int. Ed., 2003, 115, 4270 CrossRef; (d) S. Wang, T. B. Sim, Y.-S. Kim and Y.-T. Chang, Curr. Opin. Chem. Biol., 2004, 8, 371 CrossRef CAS PubMed; (e) L. A. Mitscher, Chem. Rev., 2005, 105, 559 CrossRef CAS PubMed; (f) H. Zhao, N. Neamati, S. Sunder, H. Hong, S. Wang, G. W. A. Milne, Y. Pommier and T. R. Burke Jr, J. Med. Chem., 1997, 40, 937 CrossRef CAS PubMed.
- (a) L. A. VanderVeen, A. Druckova, J. N. Riggins, J. L. Sorrells, F. P. Guengerich and L. J. Marnett, Biochemistry, 2005, 44, 5024 CrossRef CAS PubMed; (b) D. J. Wright, W. E. Jack and P. Modrich, J. Biol. Chem., 1999, 274, 31896 CrossRef CAS PubMed; (c) K. Hiramatsu, H. Miura, S. Kamei, K. Iwasaki and M. Kawakita, J. Biochem., 1998, 124, 231 CrossRef CAS; (d) C.-H. Leung, D. S.-H. Chan, B. Y.-W. Man, C.-J. Wang, W. Lam, Y.-C. Cheng, W.-F. Fong, W.-L. W. Hsiao and D.-L. Ma, Anal. Chem., 2011, 83, 463 CrossRef CAS PubMed.
- (a) X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 119, 3538 CrossRef; (b) G. Song, C. Chen, J. Ren and X. Qu, ACS Nano, 2009, 3, 1183 CrossRef CAS PubMed; (c) L.-J. Ou, P.-Y. Jin, X. Chu, J.-H. Jiang and R.-Q. Yu, Anal. Chem., 2010, 82, 6015 CrossRef CAS PubMed; (d) J. Deng, Y. Jin, G. Z. Chen and L. Wang, Analyst, 2012, 137, 1713 RSC; (e) Z. X. Zhou, Y. Du, L. B. Zhang and S. J. Dong, Biosens. Bioelectron., 2012, 34, 100 CrossRef CAS PubMed; (f) L. H. Lu, D. S. Chan, D. W. Kwong, H.-Z. He, C.-H. Leung and D.-L. Ma, Chem. Sci., 2014 10.1039/c4sc02032d; (g) Y. Huang, S. L. Zhao, M. Shi, J. Chen, Z.-F. Chen and H. Liang, Anal. Chem., 2011, 83, 8913 CrossRef CAS PubMed; (h) Q. Zhang and D.-M. Kong, Analyst, 2013, 138, 6437 RSC; (i) C. Song, G.-Y. Wang, Y.-L. Wang, D.-M. Kong, Y.-J. Wang, Y. Li and W.-J. Ruan, Chem. Commun., 2014, 50, 11177 RSC; (j) X.-P. Wang, B.-C. Yin and B.-C. Ye, RSC Adv., 2013, 3, 8633 RSC; (k) N. Wang, D.-M. Kong and H.-X. Shen, Chem. Commun., 2011, 47, 1728 RSC; (l) Y. He, B. Jiao and H. Tang, RSC Adv., 2014, 4, 18294 RSC.
- (a) K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. Colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2009, 48, 856 CrossRef CAS PubMed; (b) C. Song, G.-Y. Wang, H.-Z. Wang, Y.-J. Wang and D.-M. Kong, J. Mater. Chem. B, 2014, 2, 1549 RSC.
- (a) Y. V. Gerasimova and D. M. Kolpashchikov, Chem. Soc. Rev., 2014, 43, 6405 RSC; (b) W. B. Zhao, Z. Qin, C. S. Zhang, M. P. Zhao and H. Luo, Chem. Commun., 2014, 50, 9846 RSC; (c) F. Xuan, X. T. Luo and I. Hsing, Anal. Chem., 2012, 84, 5216 CrossRef CAS PubMed.
- Q. Xue, G. Zhang, L. Wang and W. Jiang, Analyst, 2014, 139, 3167 RSC.
- J. Li, H. E. Fu, L. J. Wu, A. X. Zheng, G. N. Chen and H. H. Yang, Anal. Chem., 2012, 84, 5309 CrossRef CAS PubMed.
- (a) H. Jia, Z. Wang, C. Wang, L. Chang and Z. Li, RSC Adv., 2014, 4, 9439 RSC; (b) F. Xuan, X. T. Luo and I. Hsing, Anal. Chem., 2013, 85, 4586 CrossRef CAS PubMed.
- (a) W. Cheng, F. Yan, L. Ding, H. Ju and Y. Yin, Anal. Chem., 2010, 82, 3337 CrossRef CAS PubMed; (b) J. Lee, K. Icoz, A. Roberts, A. D. Ellington and C. A. Savran, Anal. Chem., 2010, 82, 197 CrossRef CAS PubMed.
- (a) C. C. Richardson, I. R. Lehman and A. Kornberg, J. Biol. Chem., 1964, 239, 251 CAS; (b) S. Henikoff, Gene, 1984, 28, 351 CrossRef CAS.
- (a) Y. Huang, S. L. Zhao, Z. F. Chen, M. Shi, J. Chen and H. Liang, Chem. Commun., 2012, 48, 11877 RSC; (b) M. Luo, X. Xiang, D. S. Xiang, S. Yang, X. H. Ji and Z. K. He, Chem. Commun., 2012, 48, 7416 RSC; (c) Y. Wei, W. Zhou, Y. Xu, Y. Xiang, R. Yuan and Y. Chai, RSC Adv., 2014, 4, 39082 RSC.
- X. Zuo, F. Xia, Y. Xiao and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1816 CrossRef CAS PubMed.
- (a) A. Jeltsch, J. Alves, H. Wolfes, G. Maass and A. Pingoud, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8499 CrossRef CAS; (b) D. Li, B. Shlyahovsky, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2007, 129, 5804 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09676b |
| This journal is © The Royal Society of Chemistry 2014 |