
Isophthalonitrile (IPN) hydrogenation over K modified Ni–Co supported catalysts: catalyst characterization and performance evaluation†
Chang Liu and
Tiefeng Wang*
Beijing Key Laboratory of Green Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing, 100084, China. E-mail: wangtf@tsinghua.edu.cn; Tel: +86-10-62794132
First published on 18th November 2014
Abstract
The hydrogenation of isophthalonitrile (IPN) to meta-xylylenediamine (m-XDA) is usually catalyzed by the Raney or supported Ni based catalysts in the presence of basic additive. Although the supported catalysts are safer than the Raney Ni catalysts, they have lower selectivity to m-XDA. This work revealed that the acid sites of NiCo/Al2O3 were responsible for the condensation reactions between amines and imines, which were the dominant side reactions. Besides the original acid sites on γ-Al2O3, the loading of Ni–Co introduced new acid sites, which had a greater contribution to the condensation reactions. The K modification significantly enhanced the selectivity to m-XDA by reducing the two kinds of acid sites. Due to the formation mechanism of new acid sites and the K modification mechanism on these sites, both the K loading and K impregnation sequence affected the catalytic performance. When 3.0 wt% K was introduced to NiCo/Al2O3 by co-impregnation (3KNiCo/Al2O3), the catalyst acidity decreased by 82%, and the selectivity to m-XDA increased from 45.5% to 99.9%. The superiority of the optimized catalyst 3KNiCo/Al2O3 was also confirmed when no basic additive was used.
1. Introduction
The hydrogenation of nitriles is an important industrial process to produce primary amines,1–3 which are widely used as solvents, pharmaceuticals, disinfectants, rubber stabilizers, textile additives, and chemical intermediates.4,5 For example, meta-xylylenediamine (m-XDA), an ideal curing agent of epoxy,5,6 is produced by catalytic hydrogenation of isophthalonitrile (IPN).5–7In the industrial process of IPN hydrogenation, Raney Ni3,8 is the most widely used catalyst and has a high selectivity to m-XDA. Other Raney catalysts, such as the Raney Co9,10 and modified Raney catalysts,5 are also used. However, due to the skeleton structures,11 the Raney catalysts have low mechanical strength, leading to a high catalyst consumption. Besides, they are flammable when exposed in air, therefore they should be used very carefully to ensure safety. In the hydrogenation of IPN, the catalysts deactivate due to the condensation reactions, which requires the regeneration ability of the catalysts. However, the Raney catalysts cannot be regenerated by the common combustion method due to their flammability. In recent years, more attentions have been paid to the supported catalysts for nitrile hydrogenation reactions, with transition metals from the VIII group as the active components, including precious metals of Pd, Pt and Ru,1,12–14 and nonprecious metals such as Ni and Co.1,6,15–17 Among all the active metals, Ni and Co have the highest selectivity to primary amines,17 and the Ni and Co based catalysts are more economical for industrial use. In the supported multimetallic catalysts, the synergy effects between the metal components contribute to the enhanced catalytic performance, and the supports provide good mechanical strength. For the supported Ni based catalysts, the metal loadings had a significant influence on the product distribution, and a low selectivity to primary amines was obtained at low metal loadings. To suppress the side reactions and get a high selectivity (>90%) to primary amines, a high Ni loading (>10 wt%) of nickel-based supported catalysts was used.1,6,15–17
The mechanism of nitrile hydrogenation has been studied by many researchers.1,2,9,18 According to these studies, the mechanism of IPN hydrogenation is shown in Scheme 1. In the sequential hydrogenation of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, the highly reactive intermediate imine is formed, which has been confirmed by the attenuated total reflection infra-red (ATR-IR) spectra.19 In addition to further hydrogenation to primary amines, the condensation reactions between imines and amines occur as dominant side reactions, which form higher amines and other oligomers,1,12 and lead to catalyst deactivation. By optimizing the catalyst composition and reaction conditions, and using basic additive, these side reactions can be effectively suppressed.4,5,17,20,21
N, the highly reactive intermediate imine is formed, which has been confirmed by the attenuated total reflection infra-red (ATR-IR) spectra.19 In addition to further hydrogenation to primary amines, the condensation reactions between imines and amines occur as dominant side reactions, which form higher amines and other oligomers,1,12 and lead to catalyst deactivation. By optimizing the catalyst composition and reaction conditions, and using basic additive, these side reactions can be effectively suppressed.4,5,17,20,21
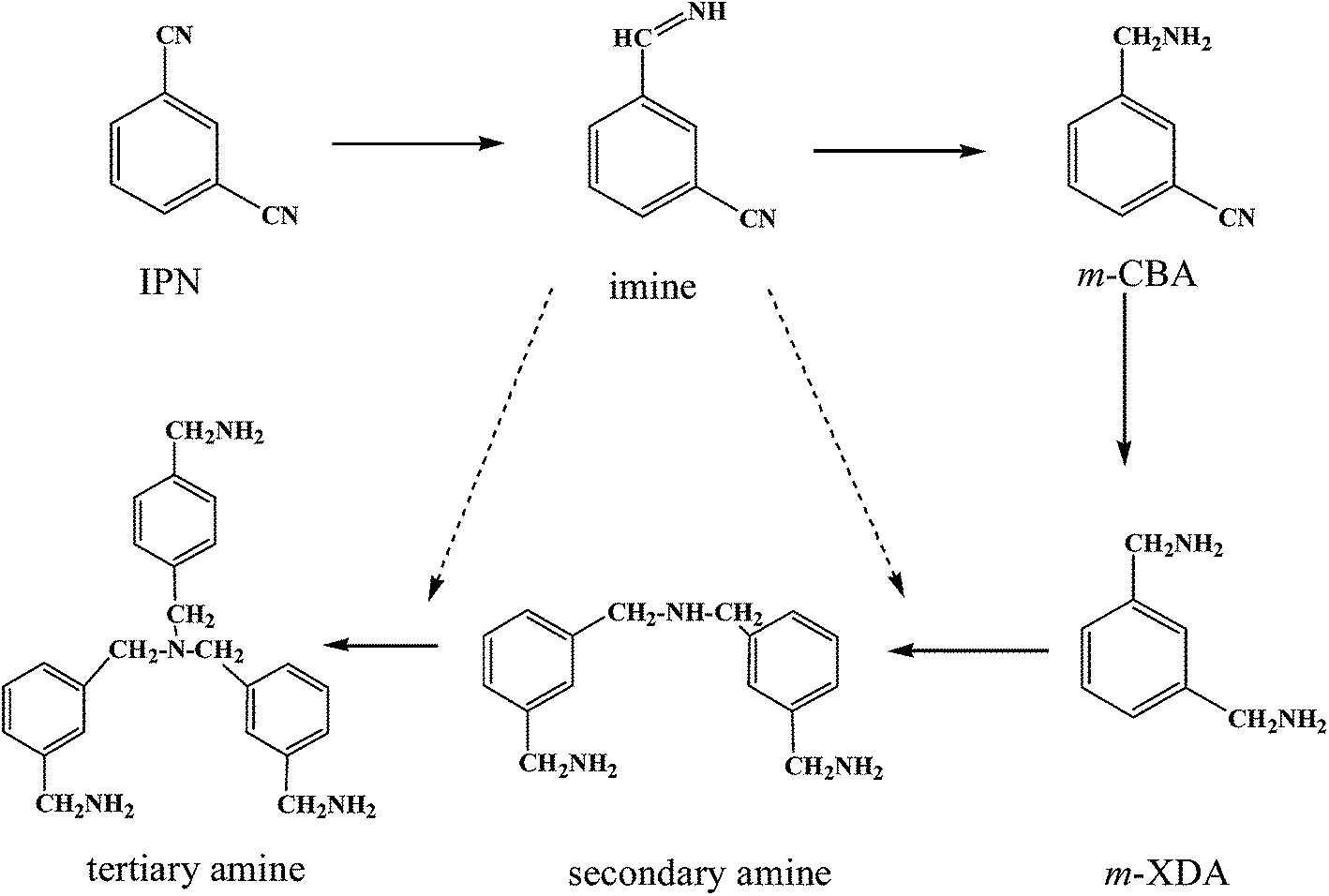 | ||
| Scheme 1 Reaction network of IPN hydrogenation reactions. | ||
In the reaction network of IPN hydrogenation, the different reactions are supposed to be catalyzed by different sites on the catalysts. A bifunctional mechanism proposed by Verhaak22 revealed that the acid sites on the catalyst were responsible for the side reactions, which was confirmed by the gas-phase hydrogenation of acetonitrile over Ni22–25 and Pt26 supported catalysts, gas-phase hydrogenation of propionitrile,27,28 and liquid-phase hydrogenation of lauronitrile.27,28 However, the underlying mechanism of the generation and effect of the acid sites is still not well understood and there are contradictory results on the effect of support acid–base property on the catalytic performance.13,29 For example, Rode et al.29 reported that in the hydrogenation of benzonitrile and acetonitrile, the supports mainly affected the Ni dispersion and only affected the catalytic activity, while the selectivity to primary amines were almost unaffected. In conclusion, it was confirmed in the literatures that the effect of support on the catalytic performance were caused by either the acid–base property or the metal–support interaction.
Alkali doping has been widely used to modify the supported catalysts. By neutralizing the acid sites, creating new basic sites, and acting as active component or electronic factor, alkali doping can enhance the catalytic activity and selectivity, and suppress the catalyst deactivation.30 A few studies have been reported on the alkali modification of the catalysts in nitrile hydrogenation.22,31 In Ni/Al2O3 catalyzed acetonitrile gas-phase hydrogenation,22 and Ni/α-Al2O3 catalyzed adiponitrile liquid-phase hydrogenation,31 K modification of the catalysts effectively increased the selectivity to primary amines. However, the systematic study on the mechanism of alkali modification based on quantitative analysis is still very limited. Besides, no relevant study on the alkali modification of the catalyst in IPN hydrogenation system has been reported.
In this work, a series of K-modified Ni–Co supported catalysts were synthesized, characterized, and evaluated in IPN hydrogenation. For easier comparison between the unmodified and modified catalysts, and studying the mechanism of K modification of the acid sites, a low Ni–Co loading (5 wt% of Ni and 1.25 wt% of Co) and a weakly acidic support (γ-Al2O3) were adopted. The effects of K loading and impregnation sequence on the catalyst acidity and catalytic performance were systematically studied. The results showed that the K modification significantly enhanced the selectivity to m-XDA by reducing the catalyst acidity.
2. Experimental
2.1. Catalyst preparation
A series of K modified Ni–Co supported catalysts, wKNiCo/Al2O3 (w is the K loading in wt%), were synthesized by incipient wetness impregnation, using nitrates (Ni(NO3)2·6H2O, 98%, Co(NO3)2·6H2O, 98.0%, KNO3, 99.0%, Alfa Aesar) as the metallic precursors, and γ-Al2O3 as the support (Alfa Aesar, grinded to 40–80 mesh). The Ni and Co loadings were 5.0 and 1.25 wt%, respectively, and the K loading varied from 0.1 to 5.0 wt% (w = 0.1–5.0). Co-impregnation was used for all K loadings. The unmodified NiCo/Al2O3 catalyst was prepared similarly, except that no K was impregnated. To study the effects of impregnation sequence, the sequential impregnation method was also used to prepare the catalysts with 1.0 and 3.0 wt% K loadings, which were denoted as wK-NiCo/Al2O3 and NiCo-wK/Al2O3 (w = 1, 3) according to the impregnation sequence. For wK-NiCo/Al2O3, K was impregnated before Ni–Co, and for NiCo-wK/Al2O3, K was impregnated after Ni–Co.The impregnated samples were aged overnight at room temperature after an ultrasonic treatment for 1 h. The catalyst precursors were then dried in air at 80 °C for 6 h, heated to 100 °C in 20 min and kept for another 20 min to remove the physically absorbed water, and then the samples were heated to 400 °C in 30 min and calcined for 4 h. Before used in the hydrogenation reactions, the catalysts were reduced in H2 flow (70 mL min−1) at 450 °C for 5 h.
2.2. Catalyst characterization
The specific surface area, pore volume and pore size distribution of the unreduced catalysts were determined by N2 adsorption–desorption with a Quantachrome autosorb iQ and AsiQwin instrument. The results were calculated by Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods.The CO-uptake of the catalysts was determined by CO chemisorption on a Quantachrome ChemBET Pulsar TPR/TPD instrument. Before the measurement, the catalysts were reduced online at 450 °C for 1 h in a 5% H2/He gas mixture at 100 mL min−1, and purged by He gas flow to remove the physically absorbed H2. The unreduced NiCo/Al2O3 and 3KNiCo/Al2O3 were observed by transmission electron microscopy (TEM, JEOL2010F) in scanning mode (STEM).
Powder X-ray diffraction (XRD) tests of the unreduced catalysts were performed using a Bruker D8 Advance powder X-ray diffractometer (40 kV, 40 mA) with a Cu Kα radiation source and a Ni filter in the 2θ range 5–90°.
The acidities of the reduced catalysts were measured by ammonia temperature-programmed desorption (NH3-TPD) using a Quantachrome ChemBET Pulsar TPR/TPD instrument. Before the TPD analysis, the catalyst of 200 mg was reduced online for 1 h at 450 °C. After the reduction, the catalyst sample was cooled to 80 °C, exposed in a 5% NH3/He gas mixture for 45 min, and purged by He gas flow to remove the physically adsorbed NH3, until the signal of thermal conductivity detector (TCD) reached a constant level. Finally, the NH3-TPD test was conducted by heating to 600 °C with the signal of NH3 desorption recorded by TCD.
Temperature programmed reduction (TPR) was also conducted on the Quantachrome ChemBET Pulsar TPR/TPD instrument. The TPR tests were carried out in a flow of a 5% H2/He mixture at 100 mL min−1 at a heating rate of 10 °C min−1. The H2 consumption was detected and recorded by TCD.
A Thermal Scientific ESCALAB 250Xi was used to examine the oxidation state of the surface atoms of the unreduced NiCo/Al2O3 and 3KNiCo/Al2O3 catalysts. The Al-Kα X-ray source was used.
2.3. Catalytic reaction
The hydrogenation reactions of IPN were carried out in a stainless steel autoclave (Weihai Chemical Machinery Co., Ltd., 250 mL) equipped with an automatic temperature control system and a magnetically driven impeller. A continuous H2 flow was controlled by a mass flow meter, and the operating pressure was controlled by a back-pressure valve at the outlet. The hydrogenation conditions were set as follows: 80 °C, 6.0 MPa, stirring speed of 800 rpm, and H2 flow rate of 190 mL min−1. Preliminary experiments showed that at these conditions the influence of external and internal diffusion was eliminated.In a typical experiment, 2.9 g of IPN (98%, J&K Chemical), 20 mL of methanol (>99.5%, Beijing Chemical Works) and 80 mL of toluene (>99.5%, Beijing Modern Oriental Fine Chemistry Co., Ltd.) were added into the reactor, and 0.086 g of NaOH (>96.0%, Beijing Chemical Works) was used as basic additive. Additional experiments without basic additive were carried out for comparison. For each experiment, 5.0 g of the pre-reduced and passivated catalyst was transferred to the solvent in the autoclave. The system was purged with H2 gas flow for 30 min under 300 rpm stirring. The reactor was heated to the reaction temperature (80 °C) under 0.3 MPa, and was then pressurized to 6.0 MPa within 5 min. At the same time, the H2 flow rate and stirring speed were set to the specified values. During the above operations, the heating process was conducted at a relatively low pressure so that only a small amount of IPN was converted during this period, and the time when the pressure reached to 6.0 MPa could be considered as zero time of the reaction.
The products were sampled online with a time interval of 10 min and were analyzed by gas chromatography (GC 7900II, Techcomp Instrument Company) equipped with a DB-1MS UI capillary column (30 m × 0.25 mm × 0.25 μm, Agilent) and an FID detector.
The conversion of IPN and the selectivity to m-XDA were calculated as:
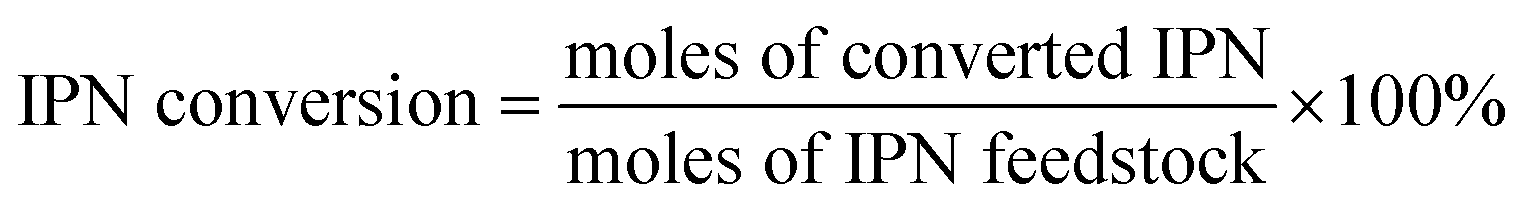 | (1) |
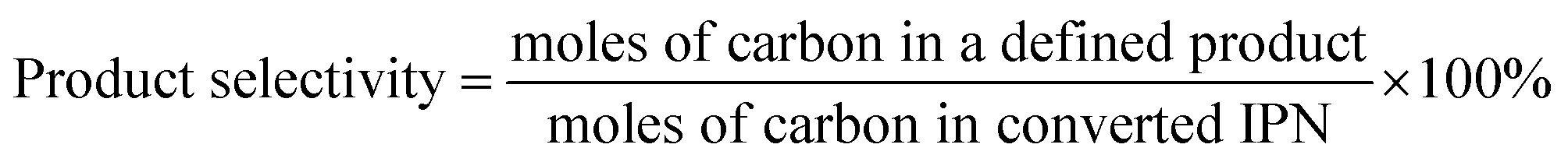 | (2) |
Besides m-XDA and unconverted IPN, the liquid sample also contained higher amines and other oligomers, which could not be detected by gas chromatography. Therefore, the species in the final liquid samples were analyzed by mass spectrometry (MS, instrument model: Q Exactive) to identify the heavier species.
3. Results and discussion
3.1. Catalyst characterization
| Catalyst | Physical properties | Acidity (mmol NH3 g−1) | Reaction resultsa | |||
|---|---|---|---|---|---|---|
| S (m2 g−1) | Vp (cm3 g−1) | Dp (nm) | kr (10−2 mol0.2 L−0.2 min−1) | Sm-XDA (%) | ||
| a Reaction conditions: 80 °C, 6.0 MPa, 5 g catalyst of 200–400 μm, 80 mL of toluene and 20 mL of methanol as solvent, 2.9 g of IPN feed, 0.086 g of NaOH, 180 mL min−1 H2 gas flow, and stirring speed of 800 rpm.b The γ-Al2O3 sample was calcined at 400 °C for 4 h before analysis.c The numbers in brackets on the left are results of wK-NiCo/Al2O3, and those in brackets on the right are results of NiCo-wK/Al2O3 (w = 1, 3). | ||||||
| γ-Al2O3b | 221 | 0.63 | 7.92 | 0.180 | — | — |
| NiCo/Al2O3 | 209 | 0.57 | 7.88 | 0.474 | 2.1 | 45.5 |
| 0.1KNiCo/Al2O3 | 209 | 0.58 | 7.92 | 0.445 | 2.0 | 50.2 |
| 0.5KNiCo/Al2O3 | 210 | 0.57 | 7.90 | 0.387 | 2.3 | 50.3 |
| 1KNiCo/Al2O3c | (205) 201 (212) | (0.65) 0.56 (0.58) | (7.86) 7.90 (7.86) | (0.167) 0.155 (0.165) | (1.9) 1.7 (1.8) | (58.6) 62.4 (59.8) |
| 2KNiCo/Al2O3 | 206 | 0.55 | 7.88 | 0.138 | 1.3 | 72.9 |
| 3KNiCo/Al2O3c | (211) 209 (209) | (0.55) 0.53 (0.56) | (7.86) 7.90 (7.84) | (0.098) 0.085 (0.100) | (2.2) 1.1 (1.6) | (79.7) 99.9 (85.4) |
| 5KNiCo/Al2O3 | 179 | 0.47 | 7.90 | 0.078 | 0.2 | 97.3 |
From theoretical calculations, the coverage of K on the support surface would reach 100% at the K loading of ≈ 5 wt%. In actual situations, however, the layer thickening and aggregation would occur at lower loading due to the non-uniform loading, and result in multilayers of K and blocking of the pore structures. Correlating to the theoretical calculation results, the above BET results could be attributed to the relatively uniform dispersion of K on the support at K loadings not higher than 3.0 wt%, which barely blocked the pore structures. Similar BET results were also reported in the literatures for Ni/SiO2, Co/SiO2 and alkali-doped (Li, Rb) Au/Al2O3.17,32 At the K loading of 5.0 wt%, the effects of K loading on the physical properties were significant, and the decrease in the BET surface area and pore volume was due to the enhanced generation of multilayers of K and the blocking of the pore structures.
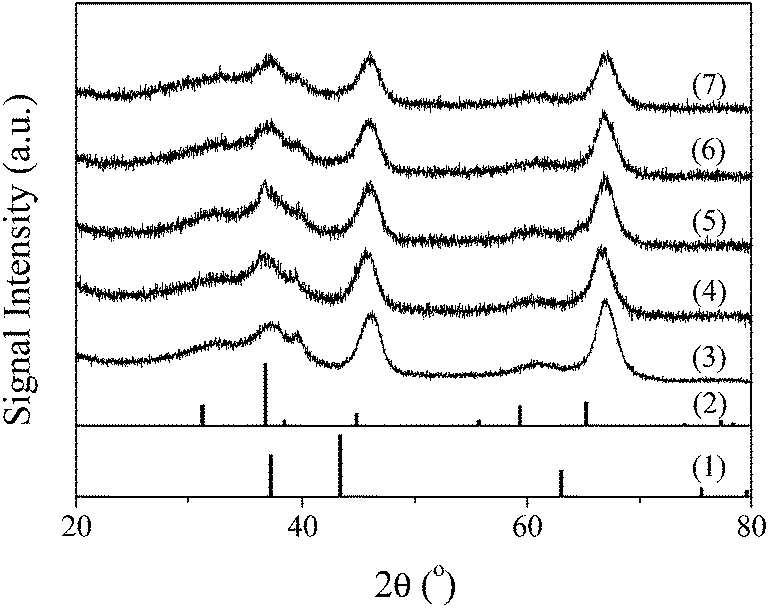 | ||
| Fig. 1 Standard spectra of (1) NiO, (2) Co3O4, and XRD results of (3) γ-Al2O3, (4) NiCo/Al2O3, (5) 0.1KNiCo/Al2O3, (6) 3KNiCo/Al2O3 and (7) 5KNiCo/Al2O3. | ||
 | ||
| Fig. 2 HAADF TEM images of (a) NiCo/Al2O3 and (b) 3KNiCo/Al2O3. | ||
 | ||
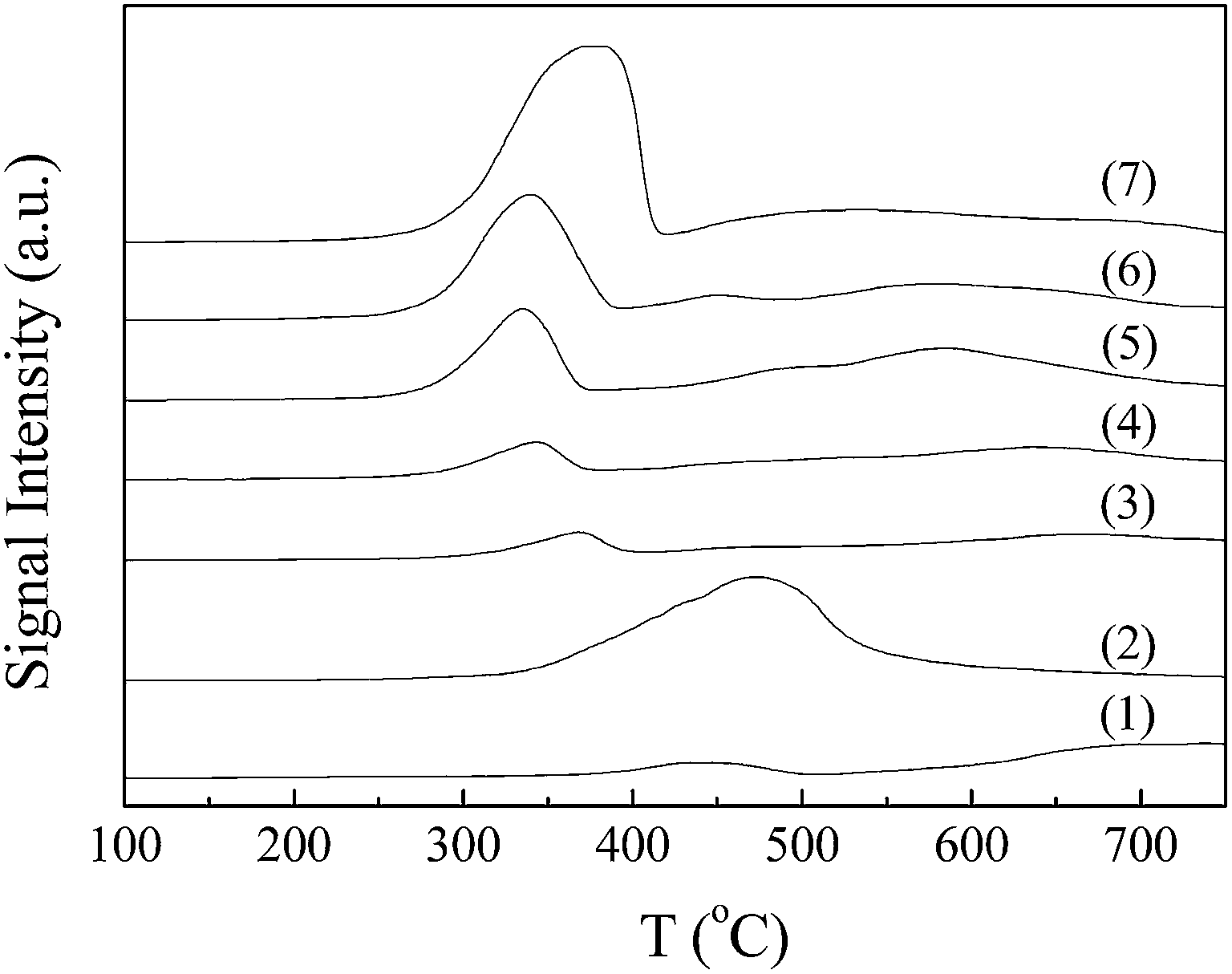
| Fig. 3 NH3-TPD profiles of (0) γ-Al2O3, (1) NiCo/Al2O3, (2) 0.1KNiCo/Al2O3, (3) 0.5KNiCo/Al2O3, (4) 1KNiCo/Al2O3, (5) 2KNiCo/Al2O3, (6) 3KNiCo/Al2O3 and (7) 5KNiCo/Al2O3. | ||
By K modification, the catalyst acidity was well modulated. With the K loading increasing from 0 to 5.0 wt%, the catalyst acidity significantly reduced from 0.474 to 0.078 mmol NH3 g−1. This could be attributed to the neutralization and blocking of the acid sites by K modification.30 Besides the K loading, the impregnation sequence of K also had an effect on the catalyst acidity. At a certain K loading (w = 1, 3), the pre-impregnated (wK-NiCo/Al2O3) and post-impregnated (NiCo-wK/Al2O3) catalysts showed similar acidity, which were higher than that of the co-impregnated catalyst (wKNiCo/Al2O3). For instance, at 3.0 wt% K loading, the catalysts prepared by sequential impregnation (3K-NiCo/Al2O3 and NiCo-3K/Al2O3) had 17.6% higher acidity than the co-impregnated catalyst 3KNiCo/Al2O3 (0.098, 0.100 and 0.085 NH3 g−1, respectively). For the catalysts modified by 1.0 wt% K, the effect of impregnation sequence was weaker than that for thier 3 wt% K modified counterparts.
 | ||
| Fig. 4 H2-TPR profiles of (1) 5 wt% Ni/Al2O3, (2) 5 wt% Co/Al2O3, (3) NiCo/Al2O3, (4) 0.1KNiCo/Al2O3, (5) 1KNiCo/Al2O3, (6) 3KNiCo/Al2O3 and (7) 5KNiCo/Al2O3. | ||
The H2-TPR results were instructive for the determination of reduction conditions of the catalysts. As reported by Verhaak et al.,22 the acidic nickel hydrosilicate was responsible for the acid–base properties of Ni/SiO2. The acidity and catalytic performance in acetonitrile hydrogenation of the Ni/SiO2 catalyst highly depended on the reduction temperature and time. With increasing reduction temperature and time, the degree of reduction of the catalyst was enhanced, leading to lower acidity. According to the literature22 and the H2-TPR results in this work, a high reduction temperature (450 °C) and a moderate reduction time (4 h) were used.
| XPS surface properties | NiCo/Al2O3 | 3KNiCo/Al2O3 | 3K-NiCo/Al2O3 | NiCo-3K/Al2O3 |
|---|---|---|---|---|
| Ni2p3/2 BE (eV) | 855.78 | 855.50 | 855.64 | 855.62 |
| Co2p3/2 BE (eV) | 781.18 | 780.89 | 780.88 | 781.00 |
| K2p3/2 BE (eV) | — | 293.08 | 293.05 | 293.11 |
| Ni–Co/Al ratio | 0.100 | 0.083 | 0.074 | 0.083 |
| K/Al ratio | — | 0.062 | 0.044 | 0.061 |
The introduction of K also affected the surface concentration of Ni–Co. As shown in Table 2, the Ni–Co/Al surface atomic ratio decreased from 0.100 on NiCo/Al2O3 to 0.074–0.083 on K modified catalyst due to the existence of K surface atoms. The Ni–Co and K surface composition was almost independent of the loading sequence of K, except that the surface K concentration of 3K-NiCo/Al2O3 was significantly lower than that of 3KNiCo/Al2O3 and NiCo-3K/Al2O3, because K was covered by the subsequent loading of Ni and Co.
3.2. Catalytic performance
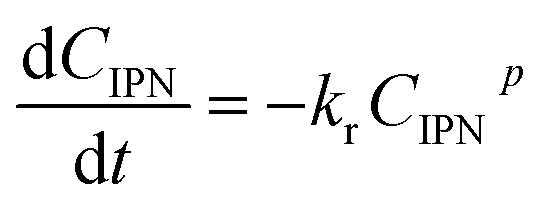 | (3) |
Using a 0.8-order reaction model, the rate constant kr could be calculated from the regression results. With the increase of K loading, kr exhibited a decreasing trend. With the K loading below 1.0 wt%, the value of kr was in the range of 2.0 × 10−2 to 2.3 × 10−2 mol0.2 L−0.2 min−1. With a further increase of the K loading, kr gradually decreased. For 2KNiCo/Al2O3 and 3KNiCo/Al2O3, the activity decreased by about 43% compared with the unmodified catalyst (kr decreased from 2.1 × 10−2 to 1.3 × 10−2 and 1.1 × 10−2 mol0.2 L−0.2 min−1). With a further increase of the K loading to 5.0 wt%, kr significantly decreased to 0.2 × 10−2 mol0.2 L−0.2 min−1. Meanwhile, the selectivity to m-XDA was much enhanced. The selectivity to m-XDA increased from 45.5% over NiCo/Al2O3 to nearly 100% over 3KNiCo/Al2O3.
For each experiment, the species in the final liquid samples were identified by MS to verify the byproducts, as shown in Fig. S4.† The MS measurement conditions were mild so that degradation of the oligomers was avoided. In the MS spectra, the peaks at m/z of 137 and 120 belonged to the protonated and deammoniated m-XDA, respectively; while those around m/z of 255 and 374 were the peaks of dimerization and trimerization products, respectively. The MS results showed that the enhanced formation of higher amines was responsible for the decreased selectivity to m-XDA. At a K loading of 3.0 wt%, only a trace amount of higher amines were detected, confirming that the condensation reactions between imines and amines were effectively suppressed by K modification.
To further analyze the effect of K modification, the selectivity to m-XDA was plotted as a function of the acidity density of the catalyst, as shown in Fig. 5. The results showed that the selectivity to m-XDA strongly depended on the catalyst acidity. The increased selectivity to m-XDA over the K modified catalysts could be attributed to the weakened adsorption of imines on the catalytic sites due to the suppressed acidity, which was the result from the blocking of the acid sites and the suppressed formation of NiAl2O4 and CoAl2O4 by K modification. The enhanced selectivity to m-XDA was also attributed to the increased electron density of Ni and Co donated by K, which favored the adsorption of nitriles by enhancing the N–metal bond, thus weakened the strength of CN or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and facilitated the attack of adsorbed H atom to the CN or CN groups in the IPN hydrogenation reaction.16 The electron-enriched Ni/Co sites also reduced the condensation reactions by inhibiting the adsorption of m-XDA, as reported in similar reactions of acetonitrile hydrogenation catalyzed by amorphous NiB and CoB alloys.44 The activity decrease in the K modified catalysts was caused by change in the morphology or component of the metal particles. Verhaak et al.22 reported that with K modification, less active Ni crystal planes were exposed on the surface, leading to the decrease in acetonitrile hydrogenation activity of Ni/Al2O3. The formation of inactive alkali metal aluminates was also a possible reason.45 In this work, the decreased activity was mainly caused by the altered surface Ni–Co composition of the catalyst. As shown by the XPS results, the Ni–Co surface density of the K modified catalysts was lower than that of NiCo/Al2O3, which was responsible for the decreased activity.
N and facilitated the attack of adsorbed H atom to the CN or CN groups in the IPN hydrogenation reaction.16 The electron-enriched Ni/Co sites also reduced the condensation reactions by inhibiting the adsorption of m-XDA, as reported in similar reactions of acetonitrile hydrogenation catalyzed by amorphous NiB and CoB alloys.44 The activity decrease in the K modified catalysts was caused by change in the morphology or component of the metal particles. Verhaak et al.22 reported that with K modification, less active Ni crystal planes were exposed on the surface, leading to the decrease in acetonitrile hydrogenation activity of Ni/Al2O3. The formation of inactive alkali metal aluminates was also a possible reason.45 In this work, the decreased activity was mainly caused by the altered surface Ni–Co composition of the catalyst. As shown by the XPS results, the Ni–Co surface density of the K modified catalysts was lower than that of NiCo/Al2O3, which was responsible for the decreased activity.
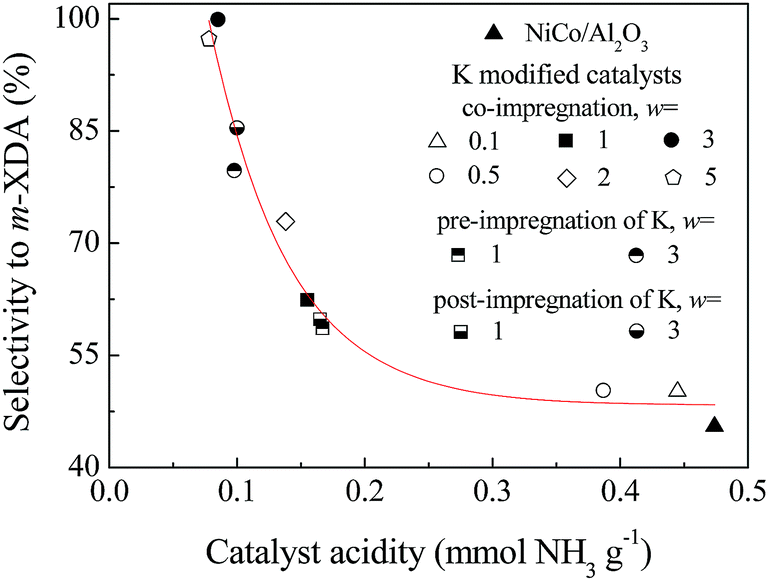 | ||
| Fig. 5 Correlation of the selectivity to m-XDA to the acidity of the wKNiCo/Al2O3 and NiCo/Al2O3 catalysts. | ||
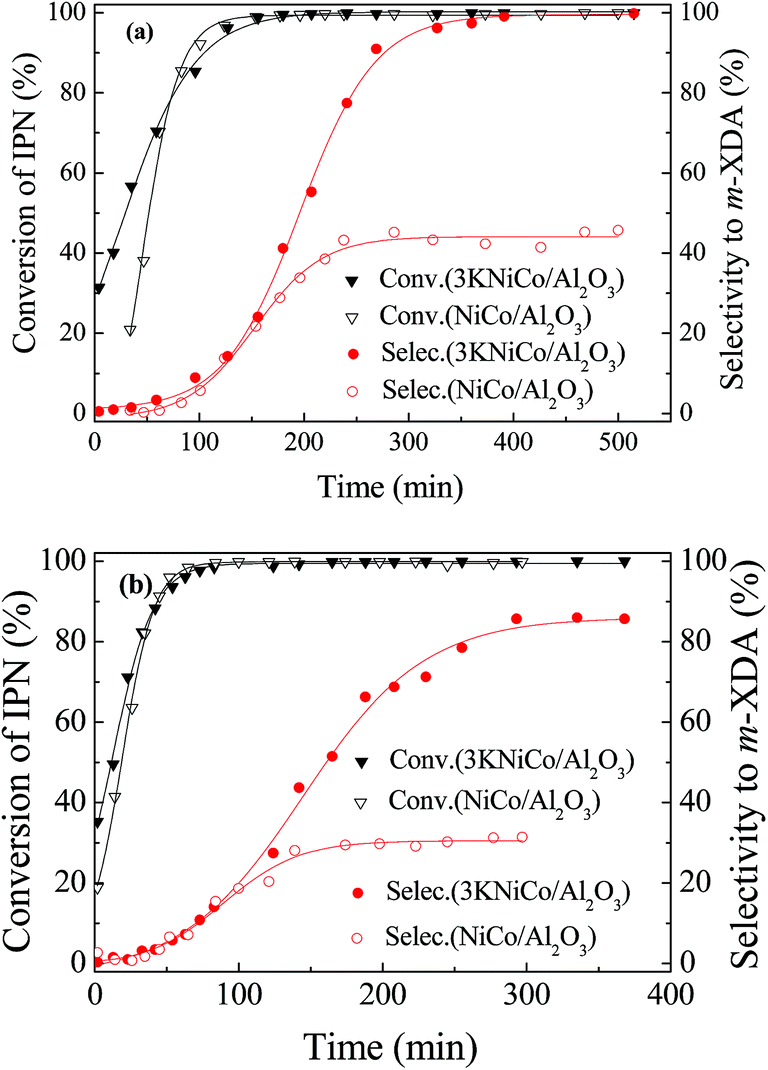 | ||
| Fig. 6 Comparison of catalytic performance of NiCo/Al2O3 and 3KNiCo/Al2O3 catalysts with or without basic additives (a) with 0.086 g NaOH as basic additive and (b) without basic additive. Other reaction conditions are the same as in Table 1. | ||
As reported in the literature,37,38 bare γ-Al2O3 had a certain amount of original acidity. In this work, it was revealed that newly formed acid sites were generated on NiCo/Al2O3 due to the formation of NiAl2O4 and CoAl2O4. Both the original and the newly formed acid sites could be covered by K modification, because both the pre- and post-impregnation of K reduced the catalyst acidity. The newly formed acid sites could be suppressed by K in the process of their formation, thus a more effective modification of acid sites was achieved by co-impregnation of K. Tennison46 proposed that there were three possible locations of the alkali metals on the alkali modified supported metallic catalysts: (1) within the crystallites of metal, possibly as a complex; (2) in contact with the support and metal simultaneously (the “hot ring” promotion); (3) on the surface of the metal or support. In this work, reasonable explanations of the modification effects of K to the NiCo/Al2O3 catalyst were as follows:
(1) The neutralization effect of K acted on both the original and newly formed acid sites, by adsorbing on the surface of γ-Al2O3 and the frontiers between Ni–Co and γ-Al2O3, leading to the reduced acidity of the wKNiCo/Al2O3 catalysts;
(2) By the “hot ring” promotion, the modification by K reduced the formation of NiAl2O4 and CoAl2O4 spinel, and suppressed the generation of new acid sites.
The discrimination between the two kinds of acid sites was reflected in the catalytic performances. In spite of their similar acidity amounts, the NiCo-wK/Al2O3 catalysts were more selective to m-XDA than wK-NiCo/Al2O3, especially for the 3.0 wt% K modified catalysts. These results indicated that the newly formed acid sites had greater contributions to the side reactions, and the K modification had different effects on the two kinds of acid sites. For the co-impregnated catalysts wKNiCo/Al2O3, the newly formed acid sites were most effectively eliminated. For the wK-NiCo/Al2O3 catalysts, the K modification mainly affected the original acid sites on the γ-Al2O3 support; while for the NiCo-wK/Al2O3 catalysts, the newly formed acid sites had been generated before the K modification, and the subsequent loading of K could only eliminate part of these acid sites. Among all the catalysts, 3KNiCo/Al2O3 prepared by co-impregnation had the most effective suppression of the acidity by K modification, and had the highest selectivity to m-XDA with only a slight decrease in the activity. In our research, a series of wKNiCo/SiO2 catalysts with different K loadings were also studied (Table S1†), the results of which further confirmed the generation mechanism and the effect of the acid sites.
| Times of reaction | Reaction resultsa | |
|---|---|---|
| kr (10−2 mol0.2 L−0.2 min−1) | Selectivity to m-XDA (%) | |
| a Reaction conditions: 80 °C, 6.0 MPa, 5 g catalyst of 200–400 μm, 80 mL of toluene and 20 mL of methanol as solvent, 2.9 g of IPN feed, without basic additives, 180 mL min−1 H2 gas flow, and stirring speed of 800 rpm.b The used catalyst was calcined in air at 400 °C for 4 h to remove the coking, and reduced in H2 for 5 h before reaction. | ||
| 1 | 2.4 | 85.7 |
| 2 | 2.1 | 86.2 |
| 3 | 2.2 | 73.2 |
| After regenerationb | 2.6 | 87.4 |
The results confirmed that the decrease of the catalyst selectivity was more significant at lower m-XDA selectivity, caused by the condensation reactions. The 3KNiCo/Al2O3 catalyst had acceptable recyclability and excellent regeneration ability even at severe reaction conditions.
4. Conclusions
The K modified Ni–Co supported catalysts, wKNiCo/Al2O3, were synthesized and evaluated for the IPN hydrogenation reaction. By K modification, the physical morphology of wKNiCo/Al2O3 was almost unchanged and the reducibility of Ni/Co oxides was enhanced except for 5KNiCo/Al2O3. The K modification decreased the total amount of the catalyst acidity and significantly enhanced the selectivity to m-XDA, with only a slight decrease in activity. Both the K loading and impregnation sequence had effects on the catalyst acidity and selectivity, indicating the original acid sites on the support and the newly formed acid sites generated from Ni–Co introduction had different effects on the reactions, and the newly formed acid sites had a greater contribution to the side reactions. Among all the catalysts, the 3KNiCo/Al2O3 catalyst prepared by co-impregnation had the best performance, having a 99.9% selectivity to m-XDA in the presence of basic additives.Acknowledgements
This work was supported by Program for New Century Excellent Talents in University of China (NCET-12-0297). This work made use of the resources of the Beijing National Center for Electron Microscopy at Tsinghua University.References
- Y. Huang and W. M. H. Sachtler, Appl. Catal., A, 1999, 182, 365–378 CrossRef CAS.
- C. De Bellefon and P. Fouilloux, Catal. Rev.: Sci. Eng., 1994, 36, 459–506 CAS.
- B. W. Hoffer and J. A. Moulijn, Appl. Catal., A, 2009, 352, 193–201 CrossRef CAS PubMed.
- R. K. Marella, K. S. Koppadi, Y. Jyothi, K. S. R. Rao and D. R. Burri, New J. Chem., 2003, 37, 3229 RSC.
- S. W. Row, T. Y. Chae, K. S. Yoo, S. D. Lee, D. W. Lee and Y. Shul, Can. J. Chem. Eng., 2007, 85, 925–928 CrossRef CAS.
- T. Shi, H. Li, L. Yao, W. Ji and C. T. Au, Appl. Catal., A, 2012, 425, 68–73 CrossRef PubMed.
- T. Y. Chae, S. W. Row, K. S. Yoo, S. D. Lee and D. W. Lee, Bull. Korean Chem. Soc., 2006, 27, 361–362 CrossRef CAS.
- F. Hochard, H. Jobic, J. Massardier and A. J. Renouprez, J. Mol. Catal. A: Chem., 1995, 95, 165–172 CrossRef CAS.
- P. Scharringer, T. Muller and J. Lercher, J. Catal., 2008, 253, 167–179 CrossRef PubMed.
- T. A. Johnson, US Pat., 5 869 653, 1999.
- B. W. Hoffer, Tuning Raney-type and Supported Ni Catalysts for Commercial Hydrogenation Reactions, 2003 Search PubMed.
- L. Hegedűs, T. Máthé and T. Kárpáti, Appl. Catal., A, 2008, 349, 40–45 CrossRef PubMed.
- Y. Huang, V. Adeeva and W. M. H. Sachtler, Appl. Catal., A, 2000, 196, 73–85 CrossRef CAS.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, J. Mol. Catal. A: Chem., 2009, 313, 74–78 CrossRef CAS PubMed.
- P. Braos-Garcıa, P. Maireles-Torres, E. Rodrıguez-Castellón and A. Jiménez-López, J. Mol. Catal. A: Chem., 2003, 193, 185–196 CrossRef.
- G. D. Yadav and M. R. Kharkara, Appl. Catal., A, 1995, 126, 115–123 CrossRef CAS.
- D. J. Segobia, A. F. Trasarti and C. R. Apesteguía, Appl. Catal., A, 2012, 445, 69–75 CrossRef PubMed.
- J. V. Braun, G. Blessing and F. Zobel, Ber. Dtsch. Chem. Ges. A, 1923, 56, 1988–2001 CrossRef.
- I. Ortiz-Hernandez and C. T. Williams, Langmuir, 2007, 23, 3172–3178 CrossRef CAS PubMed.
- Y. Huang and M. M. H. Sachtler, J. Catal., 1999, 188, 215–225 CrossRef CAS.
- Z. Cheng, Q. Wu, J. Li and Q. Zhu, Catal. Today, 1996, 30, 147–155 CrossRef CAS.
- M. J. F. M. Verhaak, Catal. Lett., 1994, 26, 37–53 CrossRef CAS.
- A. C. Gluhoi, P. Mărginean and U. Stănescu, Appl. Catal., A, 2005, 294, 208–214 CrossRef CAS PubMed.
- A. Infantesmolina, J. Catal., 2004, 225, 479–488 CrossRef CAS PubMed.
- F. M. Cabello, D. Tichit, B. Coq, A. Vaccari and N. T. Dung, J. Catal., 1997, 167, 142–152 CrossRef CAS.
- C. Park and M. A. Keane, J. Colloid Interface Sci., 2003, 266, 183–194 CrossRef CAS.
- J. B. Branco, D. Ballivet-Tkatchenko and A. P. de Matos, J. Mol. Catal. A: Chem., 2009, 307, 37–42 CrossRef CAS PubMed.
- H. Chen, M. Xue, S. Hu and J. Shen, Chem. Eng. J., 2012, 181, 677–684 CrossRef PubMed.
- C. V. Rode, M. Arai, M. Shirai and Y. Nishiyama, Appl. Catal., A, 1997, 148, 405–413 CrossRef CAS.
- W. D. Mross, Catal. Rev.: Sci. Eng., 1983, 25, 591–637 CAS.
- H. Liao, S. Liu, F. Hao, P. Liu, K. You, D. Liu and H. A. Luo, React. Kinet., Mech. Catal., 2013, 109, 475–488 CrossRef CAS.
- A. Gluhoi, N. Bogdanchikova and B. Nieuwenhuys, J. Catal., 2005, 232, 96–101 CrossRef CAS PubMed.
- L. Ji, J. Lin and H. C. Zeng, J. Phys. Chem. B, 2000, 104, 1783–1790 CrossRef CAS.
- D. Xu, W. Li, H. Duan, Q. Ge and H. Xu, Catal. Lett., 2005, 102, 229–235 CrossRef CAS.
- V. L. Barrio, P. L. Arias, J. F. Cambra and M. B. Güemez, Appl. Catal., A, 2003, 248, 211–225 CrossRef CAS.
- P. De Bokx, W. Wassenberg and J. Geus, J. Catal., 1987, 104, 86–98 CrossRef CAS.
- S. Benbenek, E. Fedoryńska and P. Winiarek, React. Kinet. Catal. Lett., 1993, 51, 189–195 CrossRef CAS.
- H. Lu, H. Yin, Y. Liu, T. Jiang and L. Yu, Catal. Commun., 2008, 10, 313–316 CrossRef CAS PubMed.
- K. M. Hardiman, C. G. Cooper and A. A. Adesina, Ind. Eng. Chem. Res., 2004, 43, 6006–6013 CrossRef CAS.
- A. Godelitsas, D. Charistos and E. A. A. Tsipis, Chem.–Eur. J., 2001, 17, 3705–3721 CrossRef.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS PubMed.
- A. A. Khassin, T. M. Yurieva, V. V. Kaichev, V. I. Bukhtiyarov, A. A. Budneva, E. A. Paukshtis and V. N. Parmon, J. Mol. Catal. A: Chem., 2001, 175, 189–204 CrossRef CAS.
- C. T. Campbell and D. W. Goodman, Surf. Sci., 1982, 123, 413–426 CrossRef CAS.
- H. Li, Y. Wu, Y. Wan, J. Zhang, W. Dai and M. Qiao, Catal. Today, 2004, 93, 493–503 CrossRef PubMed.
- S. O. Soloviev, A. Y. Kapran, S. N. Orlyk and E. V. Gubareni, J. Nat. Gas Chem., 2011, 20, 184–190 CrossRef CAS.
- S. R. Tennison, in Catalytic ammonia synthesis: fundamentals and practice, ed. J. R. Jennings, Springer, 1991 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09607j |
| This journal is © The Royal Society of Chemistry 2014 |