
A general route for synthesis of N-aryl phenoxazines via copper(I)-catalyzed N-, N-, and O-arylations of 2-aminophenols†
Nan Liu,
Bo Wang,
Wenwen Chen,
Chulong Liu,
Xinyan Wang* and
Yuefei Hu*
Department of Chemistry, Tsinghua University, Beijing 100084, P. R. China. E-mail: yfh@mail.tsinghua.edu.cn; wangxinyan@mail.tsinghua.edu.cn; Fax: +86-10-62771149; Tel: +86-10-62795380
First published on 3rd October 2014
Abstract
A novel copper(I)-catalyzed tandem reaction of N- and O-arylations of 2-[N-(2-chlorophenyl)amino]phenols was developed, by which a series of structurally novel N-aryl phenoxazines were synthesized efficiently. This success owes much to the discovery of highly efficient homogeneous copper(I)-catalyzed intramolecular O-arylation of chlorobenzenes under ligand-free-like conditions. Since 2-[N-(2-chlorophenyl)amino]phenols were prepared also by copper(I)-catalyzed N-arylation of 2-aminophenols, thus a general route for efficient synthesis of N-aryl phenoxazines was established via copper(I)-catalyzed N-, N-, and O-arylations of 2-aminophenols in two steps.
Introduction
The structural unit of phenoxazine (1) has been well recognized as an electron-donor in numerous organic compounds used in the development of dye-sensitized solar cells, laser dyes, fluorescent stains and OLEDs. When its N-atom bears an electron-accepting group, a donor–acceptor structure is formed to serve as a dipolar push–pull fluorophore or chromophore. Usually, the aryl groups are employed for such purpose and therefore N-aryl phenoxazine (2) has been gaining increasing importance.1,2 As shown in Fig. 1, an OLED using 2PXZ-OXD as a green emitter was reported recently to exhibit the highest EQE among TADF-based OLEDs to date.1a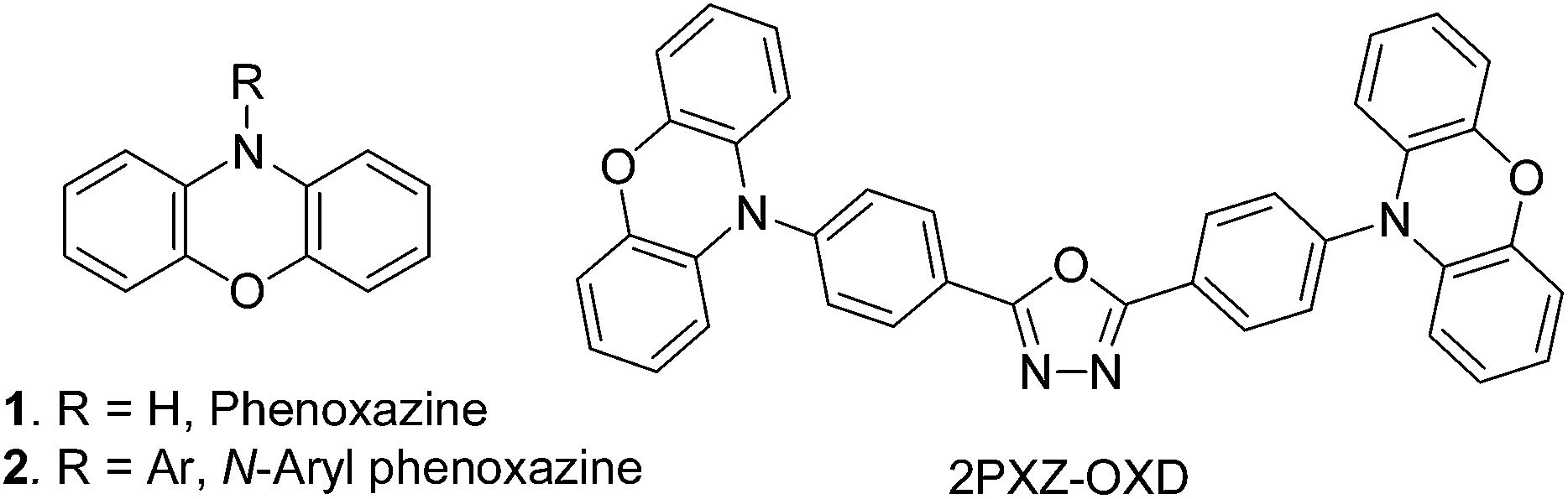 | ||
| Fig. 1 Structures of phenoxazine and N-aryl phenoxazines. | ||
Investigation showed that the construction of the skeleton of phenoxazine (1) at laboratory-scale remains a challenging task to date.3 Only a few protocols were reported for the synthesis of N-aryl phenoxazines (2) in literature, such as Pd-,1 Cu-,2,4 or base-catalyzed5 N-arylations, as well as the photocyclizations of azides.6 Despite the rapid development of Cu(I)-catalyzed N- and O-arylations in the past decade,7 none of them dealt specifically with the synthesis of N-aryl phenoxazines (2). As a result, although there are three C(Ar)–N bonds and two C(Ar)–O bonds in the molecule of 2, only the C(Ar)–N bond on C10 is usually constructed by Cu(I)-catalyzed N-arylation2 between phenoxazine (1) and halobenzenes (3). Even worse, the most often used procedure for this N-arylation was established as early as in 1957, in which the toxic nitrobenzene was used as a solvent for producing high temperature.4 Thus, it is necessary to develop a mild method for an efficient preparation of 2 to easily achieve the molecular diversity.
As shown in Scheme 1, we report herein a novel Cu(I)-catalyzed tandem reaction for N- and O-arylations of 2-[N-(2-halophenyl)amino]phenols (5), by which a series of the derivatives of 2 were prepared efficiently in one-flask. Since the precursor 5 was prepared also by Cu(I)-catalyzed N-arylation of 2-aminophenols (4), this work in fact presents a general route for efficient synthesis of 2 by Cu(I)-catalyzed N-, N-, and O-arylations of 2-aminophenols (4) in two steps.
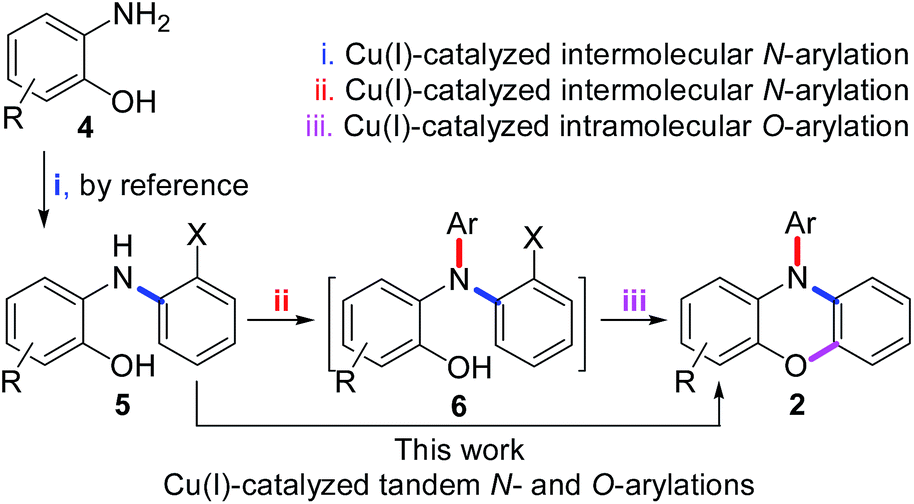 | ||
| Scheme 1 A general route for efficient synthesis of 2. | ||
Results and discussion
Due to the steric hindrance, Cu(I)-catalyzed N-arylation of diarylamine is much more difficult than that of monoarylamine. Thus, the synthesis of triarylamines usually required refluxing the mixture of the reactants, catalyst and/or ligand in high boiling solvent (toluene, NMP or DMF) in the presence of a strong base (KOtBu, NaOtBu or LiNH2).8 However, when 2-aminophenols (4) were used as the substrates, their N-arylations could proceed with weak bases under ligand-free conditions.9 As shown in Scheme 2, by using different ratios of 2-aminophenol (4a) and iodobenzene (3a), the desired diarylamine 7 or triarylamine 8 was synthesized in high yields. It has been confirmed that the compounds 4a and 7 not only were reactants, intermediates, or products, but also served as ligands. Therefore, the ligand-free conditions for these N-arylations can be considered as ligand-free-like conditions. So far, only the derivatives of 8 bearing two identical aryl groups were prepared by this method. | ||
| Scheme 2 Ligand-free-like N-arylations of 4a. | ||
When we repeated the procedure for the synthesis of 8 starting from 4a, we found that the yield of the intermediate 7 remained in less than 3% during the entire process. It was clearly revealed that the conversion of 4a into 7 was the rate-determining step and the conversion of 7 into 8 was a fast process. As shown in Scheme 3, this hypothesis was proved by using the pre-made 7 as a substrate to give 8 in 88% yield within 4 h.
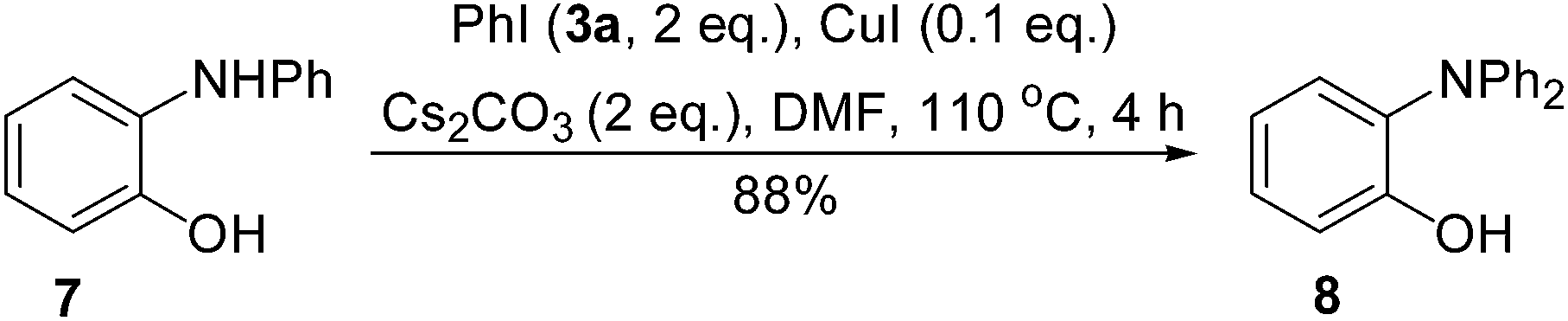 | ||
| Scheme 3 A fast conversion of 7 into 8. | ||
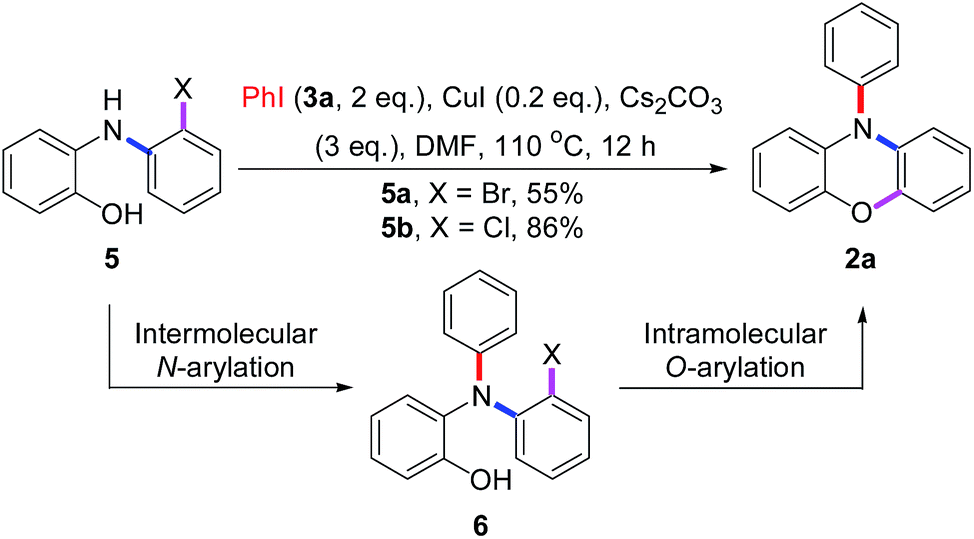
This result also strongly indicated that the unsymmetric triphenylamine 2-[N-(2-halophenyl)-N-phenylamino]phenol (6) may be synthesized easily via Cu(I)-catalyzed N-arylation between iodobenzene (3a) and 2-[N-(2-halophenyl)amino]phenol (5). Thus, we may expect that N-phenyl phenoxazine (2a) is synthesized via Cu(I)-catalyzed intramolecular O-arylation of 6. To our surprise, a novel tandem reaction for N- and O-arylations of 5 occurred to yield 2a directly instead of the excepted 6 when the mixture of 3a and 5 was treated with CuI (Scheme 4). As shown in Fig. 2, the structure of 2a was confirmed by single crystal X-ray diffraction analysis.
 | ||
| Scheme 4 The tandem reaction for N- and O-arylations of 5. | ||
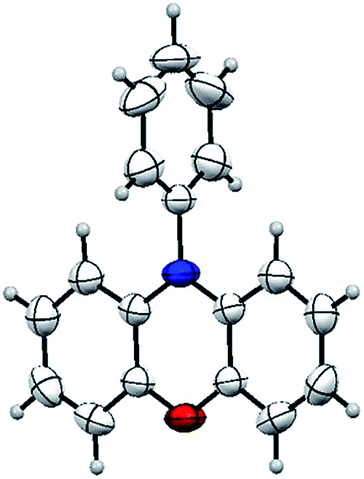 | ||
| Fig. 2 The structure of 2a. | ||
To further understand the results in Scheme 4, the pre-made compounds 6a and 6b were tested as starting materials. As shown in Scheme 5, both of them carried out Cu(I)-catalyzed O-arylations smoothly to give 2a in 95% yields. Thus, two conclusions were drawn: first, the compound 6 was the intermediate for the tandem reaction; secondly, the problem that 5a gave the lower yield of 2a in the tandem reaction occurred in the conversion of 5a into 6a, in which the highly reactive bromide group may carry out an undesired N-arylation between two molecules of 5a, besides the desired N-arylation between 5a and 3a.
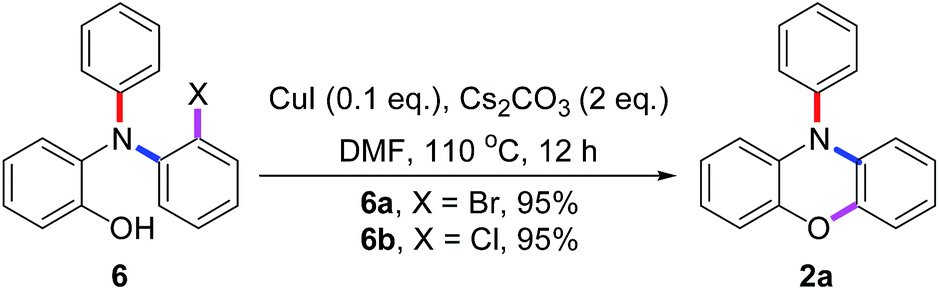 | ||
| Scheme 5 Cu(I)-catalyzed intramolecular O-arylations of 6a and 6b. | ||
It was well known that Cu(I)-catalyzed O-arylation of chloro-benzenes was the most difficult task compared with that of bromo- and iodobenzenes. Only a few successful procedures were reported in literature, such as by using heterogeneous nano-catalysts10a–c or large amounts of ligands (0.2–0.8 equiv.).10d,e To the best of our knowledge, the conversion of 6b into 2a is the first example of highly efficient homogeneous Cu(I)-catalyzed O-arylation of chlorobenzene under the ligand-free-like conditions. This work is so important because the O-arylation by using chloroaromatics as arylating reagents to replace bromo- or iodoaromatics in the synthesis of aryl ethers has been identified to be one of “dream reactions” by the ACS-GCI Pharmaceutical Roundtable in 2005.11
Therefore, we were encouraged to study the O-arylation of chlorobenzene further. As shown in Scheme 6, no intermolecular O-arylation product 9 was obtained at all from the substrate 8 and chlorobenzene under ligand-free-like conditions. Very low yields of 9 were obtained with the ligands L1–L5 (the most efficient ligands reported in literature).10d,e,12 In a recent reference, the synthesis of xanthones by using Cu(I)-catalyzed intramolecular O-arylation of chlorobenzenes was reported to fail,13 even though the corresponding bromo- and iodobenzenes worked well. Therefore, we strongly believed that the highly efficient formation of 2a from 6b may depend on the structural nature of 6b rather than the differences between the intermolecular and intramolecular O-arylations.
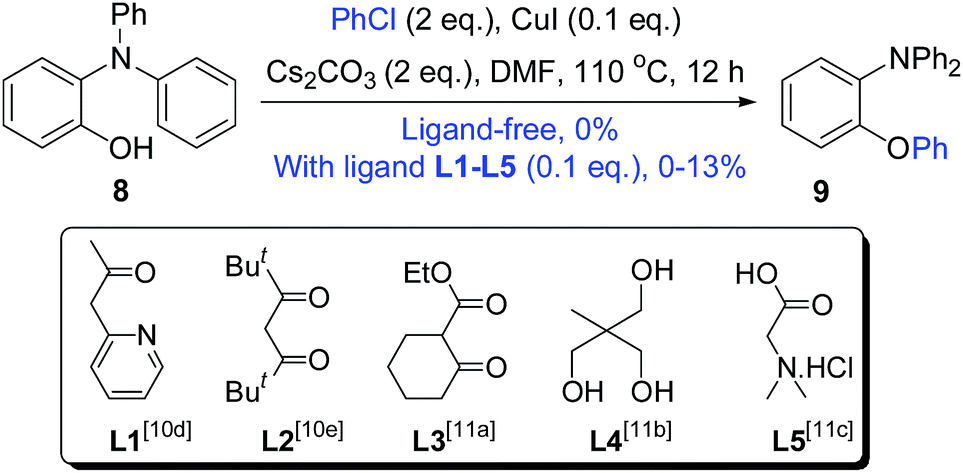 | ||
| Scheme 6 Cu(I)-catalyzed intermolecular O-arylations of 8 and PhCl. | ||
As shown in Scheme 7, when 5b was treated with CuI in the absence of PhI (3a), it was recovered in 93% yield without any intramolecular O-arylated product phenoxazine (1). Thus, we hypothesized that the Cu(I)-catalyzed O-arylations of 5b, 6b and 8 may be mainly controlled by the electronic effect rather than by the steric effect. Those phenomena may arise from the fact that 5b, 6b and 8 are also redox-active ligands, which may have different abilities to store and release electrons during the catalytic reactions,14 but how remains unknown.
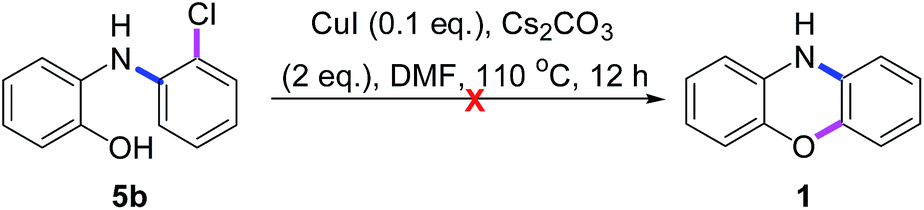 | ||
| Scheme 7 Cu(I)-catalyzed intramolecular O-arylations of 5b. | ||
Next, the reaction solvents and copper-resources were screened by using the conversion of 5b into 2a as a model reaction. As shown in Table 1, the best result was still obtained when the amounts of PhI (3a) and CuI were reduced as low as 1.1 equiv. and 0.05 equiv, respectively, in n-butylnitrile (entry 3). But, all other solvents gave relatively lower yields of 2a (entries 6–13). Although CuI, CuCl, and CuCN (entries 3, 14 and 15) gave comparable yields of 2a, we preferred to choose CuI for its chemical stability and easy performance.
|
|||
|---|---|---|---|
| Entry | [Cu]-resource (eq.) | Solvent | 2ab (%) |
| a The mixture of 5b (1 mmol), 3a, [Cu] and Cs2CO3 in solvent (2 mL) in a Schlenk tube was heated under N2.b Isolated yields were obtained. | |||
| 1 | CuI (0.2) | n-PrCN | 96 |
| 2 | CuI (0.1) | n-PrCN | 96 |
| 3 | CuI (0.05) | n-PrCN | 96 |
| 4 | CuI (0.04) | n-PrCN | 90 |
| 5 | CuI (0.00) | n-PrCN | 6 |
| 6 | CuI (0.05) | DMF | 86 |
| 7 | CuI (0.05) | PhMe | 83 |
| 8 | CuI (0.05) | 1,4-Dioxane | 52 |
| 9 | CuI (0.05) | (CH2OH)2 | 49 |
| 10 | CuI (0.05) | (CH2Cl)2 | 20 |
| 11 | CuI (0.05) | MeCN | 12 |
| 12 | CuI (0.05) | THF | 10 |
| 13 | CuI (0.05) | MeOH | Trace |
| 14 | CuCl (0.05) | n-PrCN | 95 |
| 15 | CuCN (0.05) | n-PrCN | 92 |
| 16 | Cu(OAc)2·H2O (0.05) | n-PrCN | 82 |
| 17 | Cu2O (0.05) | n-PrCN | 75 |
| 18 | CuBr (0.05) | n-PrCN | 73 |
| 19 | Cu(CO2)2·4H2O (0.05) | n-PrCN | 63 |
| 20 | CuF2·H2O (0.05) | n-PrCN | 31 |
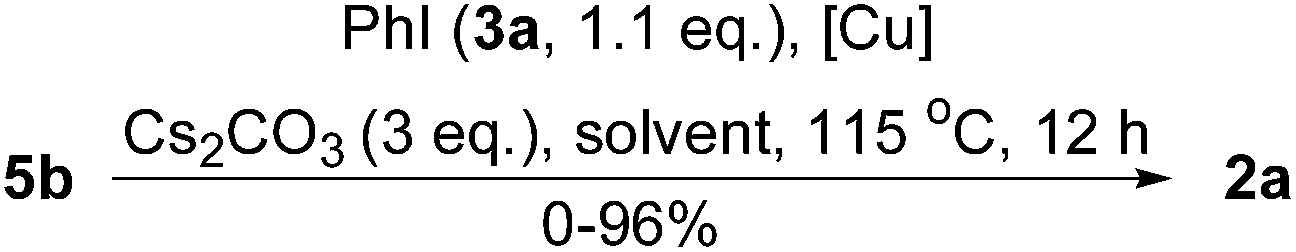
Then, the effects of reaction time and bases were tested. As shown in Table 2, the yield of 2a was increased by increasing the reaction time (entries 1–3). Both Cs2CO3 and K3PO4 (entry 4) were suitable bases for this reaction, but all others were inactive (entries 5–8). Finally, the entry 3 was assigned as our standard conditions.
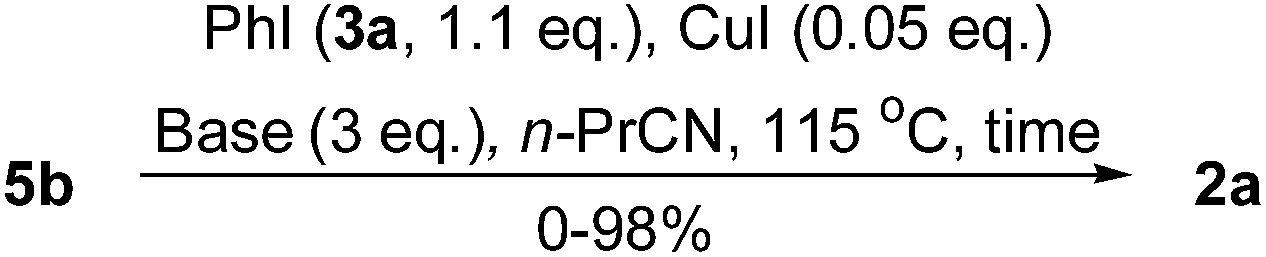
To generalize this novel method, the substrate scope was tested. As shown in Scheme 8, all desired products 2a–2x were obtained in good to excellent yields and some of them (2a, 2d, 2e, and 2i) were obtained in almost quantitative yields. However, the iodobenzenes substituted by –Br, –NO2 and –CN could not give the satisfactory yields of products (2j–2l and 2q, 2r) under the standard conditions. It may be caused by the fact that 4-bromo-iodobenzene has two reaction sites and both of them could carry out the Cu(I)-catalyzed N-arylations. The –NO2 or –CN substituted iodobenzenes may have high reactivity to carry out both Cu(I)-catalyzed N- and O-arylations simultaneously with the substrates. But, these problems could be solved easily by heating the reaction mixture at 65 °C for the first 6 h to finish the N-arylation and then at 115 °C for another 18 h to finish the O-arylation. Unfortunately, dissatisfactory yields of 2a (34%), 2d (23%), and 2q (70%) were obtained when bromobenzene, 4-bromo-toluene, and 4-bromo-nitrobenzene were used as the N-arylating reagents.
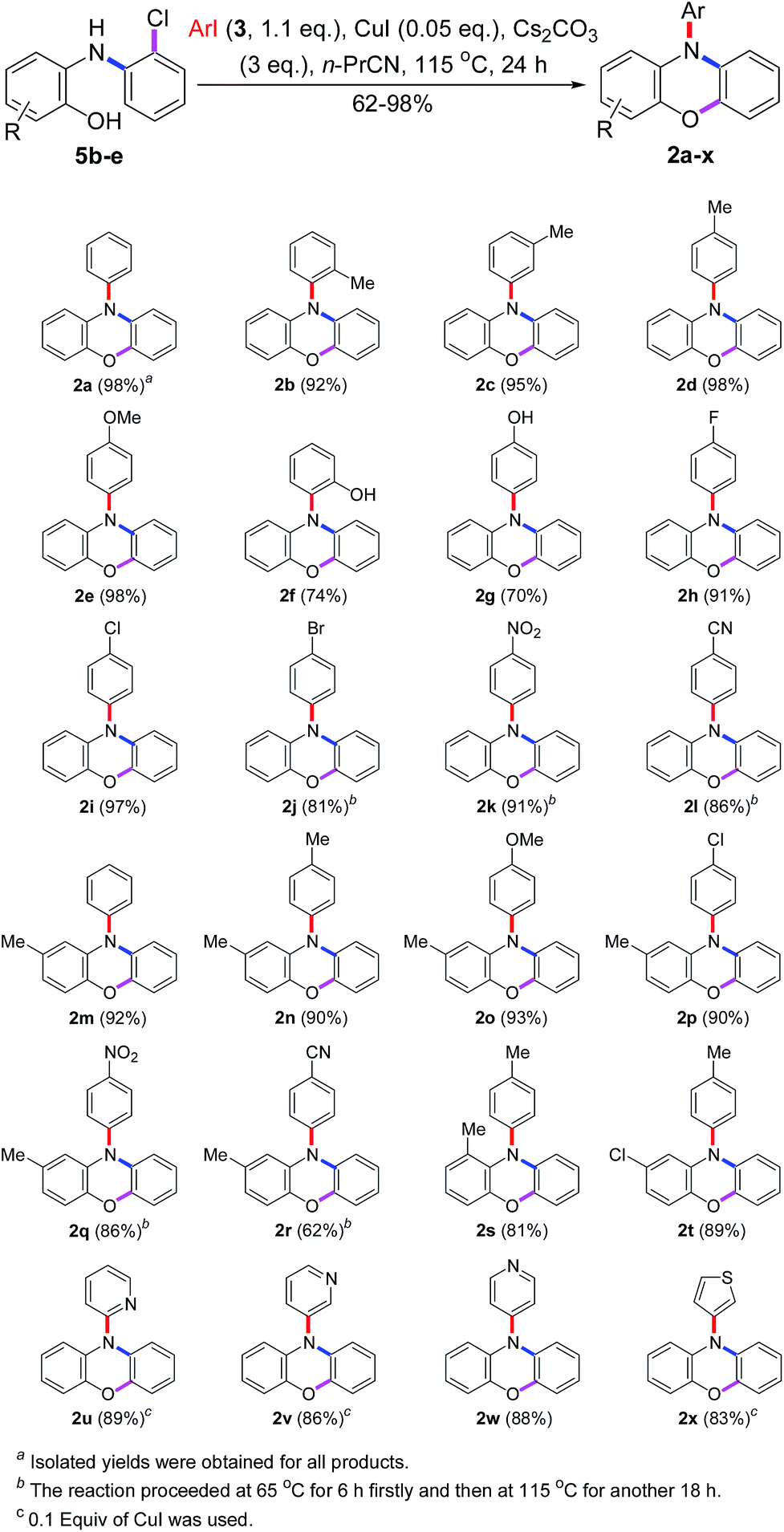 | ||
| Scheme 8 Substrate scope of the tandem reaction. | ||
Conclusions
A novel Cu(I)-catalyzed tandem reaction for N- and O-arylations of 2-[N-(2-chlorophenyl)amino]phenols was developed, by which a series of N-phenyl phenoxazines were prepared efficiently in “one-pot”. This work not only provides a general route for efficient synthesis of N-phenyl phenoxazines from the commercially available 2-aminophenols in two steps, but also presents an interesting example to construct the complicated molecules entirely by Cu(I)-catalyzed arylations from the simple starting materials.Experimental
General information
All melting points were determined on a Yanaco melting point apparatus and were uncorrected. IR spectra were recorded as KBr pellets on a Nicolet FT-IR 5DX spectrometer. All spectra of 1H NMR (300 MHz) and 13C NMR (75 MHz) were recorded on a JEOL JNM-ECA 300 spectrometer in CDCl3 (otherwise as indicated). TMS was used as an internal reference and J values are given in Hz. HRMS were obtained on a Bruker micrOTOF-Q II spectrometer. The substituted 2-[N-(2-chlorophenyl)amino]phenols 5b (R = H), 5c (R = 4-Me), 5d (R = 3-Me) and 5e (R = 4-Cl) were prepared by the reported procedure9b (See ESI†).The similar procedure was used for the preparation of products 2b–2x.
Acknowledgements
This work was supported by the National Natural Scientific Foundation of China (no. 21372142, 21472107 and 21072112).Notes and references
- (a) J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2013, 1, 4599–4604 RSC; (b) R. Menzel, S. Kupfer, R. Mede, D. Weiß, H. Görls, H. González and R. Becker, Eur. J. Org. Chem., 2012, 5231–5247 CrossRef CAS; (c) H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC; (d) Y. Park, B. Kim, C. Lee, A. Hyun, S. Jang, J.-H. Lee, Y.-S. Gal, T. H. Kim, K.-S. Kim and J. Park, J. Phys. Chem. C, 2011, 115, 4843–4850 CrossRef CAS; (e) K. Ono, M. Joho, K. Saito, M. Tomura, Y. Matsushita, S. Naka, H. Okada and H. Onnagawa, Eur. J. Inorg. Chem., 2006, 3676–3683 CrossRef CAS; (f) G.-S. Jiao, A. Loudet, H. B. Lee, S. Kalinin, L. B.-A. Johansson and K. Burgessa, Tetrahedron, 2003, 59, 3109–3116 CrossRef CAS.
- (a) J. Sun, H. Jiang, J. Zhang, Y. Tao and R. Chen, New J. Chem., 2013, 37, 977–985 RSC; (b) K. M. Karlsson, X. Jiang, S. K. Eriksson, E. Gabrielsson, H. Rensmo, A. Hagfeldt and L. Sun, Chem.–Eur. J., 2011, 17, 6415–6424 CrossRef CAS PubMed; (c) K. Kim, S. Jeong, C. Kim, E. Kim, E. Kwon, H. Kim, B.-D. Choi and Y. S. Han, Synth. Met., 2010, 160, 549–555 CrossRef CAS PubMed; (d) Y. Zhu, A. P. Kulkarni, P.-T. Wu and S. A. Jenekhe, Chem. Mater., 2008, 20, 4200–4211 CrossRef CAS; (e) Y. Zhu, A. P. Kulkarni and S. A. Jenekhe, Chem. Mater., 2005, 17, 5225–5227 CrossRef CAS; (f) A. Higuchi and Y. Shirota, Mol. Cryst. Liq. Cryst., 1994, 242, 127–133 CrossRef.
- (a) I. Thome and C. Bolm, Org. Lett., 2012, 14, 1892–1895 CrossRef CAS PubMed , and the references cited therein; (b) P. Thansandote, E. Chong, K.-O. Feldmann and M. Lautens, J. Org. Chem., 2010, 75, 3495–3498 CrossRef CAS PubMed.
- H. Gilman and L. Moore, J. Am. Chem. Soc., 1957, 79, 3485–3487 CrossRef CAS.
- A. Mori, S. Kinoshita, M. Furusyo and K. Kamikawa, Chem. Commun., 2010, 46, 6846–6848 RSC.
- (a) L. S. Klimenko, I. A. Oskina, V. M. Vlasov, I. Y. Bagryanskaya and Y. V. Gatilov, Russ. Chem. Bull., 2007, 56, 1130–1134 CrossRef CAS PubMed; (b) L. S. Klimenko, S. Z. Kusov, V. M. Vlasov, E. N. Tchabueva and V. V. Boldyrev, Mendeleev Commun., 2006, 16, 224–225 CrossRef PubMed.
- For selected reviews, see: (a) J. Keilitz, H. A. Malik and M. Lautens, Top. Heterocycl. Chem., 2013, 32, 187–224 Search PubMed; (b) T. R. M. Rauws and B. U. W. Maes, Chem. Soc. Rev., 2012, 41, 2463–2497 RSC; (c) C. Fischer and B. Koenig, Beilstein J. Org. Chem., 2011, 7, 59–74 CAS; (d) E. Sperotto, G. P. M. van Klink, G. van Koten and G. J. de Vries, Dalton Trans., 2010, 39, 10338–10351 RSC; (e) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed; (f) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed; (g) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed.
- For selected references, see: (a) A. Tlili, F. Monnier and M. Taillefer, Chem. Commun., 2012, 48, 6408–6410 RSC; (b) C.-K. Tseng, C.-R. Lee, C.-C. Han and S.-G. Shyu, Chem.–Eur. J., 2011, 17, 2716–2723 CrossRef CAS PubMed; (c) H. Xu and C. Wolf, Chem. Commun., 2009, 1715–1717 RSC; (d) N. S. Nandurkar, M. J. Bhanushali, M. D. Bhor and B. M. Bhanage, Tetrahedron Lett., 2007, 48, 6573–6576 CrossRef CAS PubMed; (e) Y.-H. Liu, C. Chen and L.-M. Yang, Tetrahedron Lett., 2006, 47, 9275–9278 CrossRef CAS PubMed; (f) A. S. Gajare, K. Toyota, M. Yoshifuji and F. Ozawab, Chem. Commun., 2004, 1994–1995 RSC; (g) N. M. Patil, A. A. Kelkar, Z. Nabi and R. V. Chaudhari, Chem. Commun., 2003, 2460–2461 RSC; (h) A. A. Kelkar, A. M. Patil and R. V. Chaudhari, Tetrahedron Lett., 2002, 43, 7143–7146 CrossRef CAS; (i) R. K. Gujadhur, D. Venkataraman and J. T. Kintigh, Tetrahedron Lett., 2001, 42, 4791–4793 CrossRef CAS; (j) R. K. Gujadhur, C. G. Bates and D. Venkataraman, Org. Lett., 2001, 3, 4315–4317 CrossRef CAS PubMed.
- (a) Y. Li, H. Wang, L. Jiang, F. Sun, X. Fu and C. Duan, Eur. J. Org. Chem., 2010, 6967–6973 CrossRef CAS; (b) D. Maiti and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 17423–17429 CrossRef CAS PubMed; (c) H. F. Wang, Y. M. Li, F. F. Sun, Y. Feng, K. Jin and X. N. Wang, J. Org. Chem., 2008, 73, 8639–8642 CrossRef CAS PubMed.
- (a) R. Arundhathi, D. Damodara, R. R. Likhar, M. L. Kantam, P. Saravanan, T. Magdaleno and S. H. Kwon, Adv. Synth. Catal., 2011, 353, 1591–1600 CrossRef CAS; (b) B. Sreedhar, R. Arundhathi, P. L. Reddy and M. L. Kantam, J. Org. Chem., 2009, 74, 7951–7954 CrossRef CAS PubMed; (c) J. Y. Kim, J. C. Park, A. Kim, A. Y. Kim, H. J. Lee, H. Song and K. H. Park, Eur. J. Inorg. Chem., 2009, 4219–4223 CrossRef CAS; (d) Q. Zhang, D. Wang, X. Wang and K. Ding, J. Org. Chem., 2009, 74, 7187–7190 CrossRef CAS PubMed; (e) N. Xia and M. Taillefer, Chem.–Eur. J., 2008, 14, 6037–6039 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, H. L. Leazer, R. J. Linderman, Jr., K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- (a) X. Lv and W. Bao, J. Org. Chem., 2007, 72, 3863–3867 CrossRef CAS PubMed; (b) Y.-J. Chen and H.-H. Chen, Org. Lett., 2006, 8, 5609–5612 CrossRef CAS PubMed; (c) D. Ma and Q. Cai, Org. Lett., 2003, 5, 3799–3802 CrossRef CAS PubMed.
- N. Barbero, R. SanMartin and E. Dominguez, Green Chem., 2009, 11, 830–836 RSC.
- (a) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS; (b) W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2011, 50, 3356–3358 CrossRef CAS PubMed , and the references therein.
Footnote |
| † Electronic supplementary information (ESI) available: Characterizations of compounds 5b–e, 1H and 13C NMR spectra for products 2a–2x and CIF file for the single crystal X-ray diffraction analysis of 2a. CCDC 1022191. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra09593f |
| This journal is © The Royal Society of Chemistry 2014 |