
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreservation of carotenes in the deacidification of crude palm oil
C. Pirolaa,
F. Galli*a,
A. Comazzia,
F. Manentib and
C. L. Bianchia
aDip. Chimica, Università degli Studi di Milano, via C. Golgi 19, 20133 Milan, Italy. E-mail: federico.galli@unimi.it
bDip. Chimica, Materiali e Ingegneria Chimica “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milan, Italy
First published on 17th September 2014
Abstract
Carotenoids in CPO can be preserved during the deacidification processes performed under mild conditions. The carotenes adsorption on the catalyst surface makes them more stable to air oxidation and to the oil acid content. Satisfactory FFA conversions were obtained and the catalyst showed good performance after 186 hours of work.
Carotenoids are terpenoid pigments widely present in natural compounds with noteworthy biological functions. For example β-carotene, a precursor of vitamin A in humans, plays the role of antioxidant against cancer and promotes the stimulation of the immune system.1 Carotenoids are important commercial products and, being accepted as a food-grade additive, are exempt from certification.2 Another important use of carotenes is in cosmetic preparations. Their market is growing rapidly and over 90% of their commercial availability is covered by chemically synthesised carotenoids. Crude Palm Oil (CPO), largely produced in Malaysia, represents one important source of carotenoids. Its orange colour is due to the presence of carotenes that can be extracted. The conventional recovery methods are based on a solvent extraction but are time consuming and need a large use of organic solvents, expensive and not environmentally friendly.3 Another possibility is represented by molecular distillation, but this unit operation is characterized by high implant costs.4 BD is a non-toxic, biodegradable fuel.5 It is obtained by the transesterification of vegetable oils or animal fats with methanol in homogeneously-catalyzed processes.6,7
Palm oil is one of the main feedstock for biodiesel (BD) production. Indeed, it has been studied as a potential feedstock for the production of bio-fuels because of its high production particularly in the Asian countries. For what concerns the competition with the food chain Lam8 demonstrated that with proper management and strategies, the production of BD from palm oil does not threaten oils and fats global supply at least until 2019. Currently, the main problem for the commercialization of BD is its final cost, that strongly depends by the feedstock used (about 85% of the total).9 Not refined or waste oils feedstock represent a very convenient way in order to lower biodiesel production costs, but refining process are required. In particular, it is important that the total content of Free Fatty Acid (FFA) in the oil is lower than 0.5 wt% to avoid saponification reaction with the basic catalyst needed in the transesterification reaction. The recovery of carotenes from the oil should be an important way to further increase the economic value of the whole process but the conventional physical refining process, i.e. the alkali refining method,10 destroys the carotenes.3
To lower the acid content, instead, heterogeneously catalyzed esterification of FFA could be used; a remarkable advantage of heterogeneous catalysis is the easier separation and recovery of the catalyst after the reaction, together with the fact that the esterification reaction produces fatty acid methyl ester, i.e. BD, already at this preliminary step.11–14 Aim of this work was to investigate the possibility to perform the deacidification reaction of CPO preserving the stability of carotenes, thanks to the mild conditions (T < 70 °C and room pressure) required from this technology. Deacidification tests were conducted by monitoring both the FFA and carotenes content, using heterogeneous Amberlyst A46 resins15 as catalyst, kindly provided by Dow Chemicals Company. This catalyst is a copolymer of styrene and divinyl benzene showing sulphonic (–SO3H) groups as active sites. The main characteristics of this resin are summarized in Table 1. This particular sample was chosen because of its peculiar properties, in fact, the active sites are located only at the catalyst surface and not in the internal pores. Using A46 all the internal diffusion phenomena of reactant and products could be neglected, as already discussed in a previous paper.14 We chose to use a limited amount of methanol in the reacting mixture, because, differently from several papers,16–18 our purpose is to avoid the formation of a double liquid phase due to the low solubility of methanol in oil. This situation is advantageous because prevents both FFA extraction and limits the diffusion phenomena.19,20
| Catalyst | Surface area (m2 g−1) | Average pore diameter (Å) | Total pore volume (mL g−1) | Acidity (meq. H+ per g) | Max working temperature (°C) |
|---|---|---|---|---|---|
| Amberlyst 46 | 75 | 235 | 15 | >0.4 | 120 |
Before the use, the resin was dried at 80 °C in an oven for 14–16 h. Higher temperatures are not allowed due to the risk of losing sulphonic acid sites (desulfurization of the catalyst polystyrene matrix).
The exchange capacity of the catalyst in its wet form was evaluated by total exchange with sodium chloride solution and subsequent titration.12 A value of 0.43 ± 0.01 meq. g−1 was obtained which is consistent with the manufacturer data.
All the experimental data were gathered using a 3-necked 500 mL batch reactor, equipped with a thermometer, a reflux condenser to avoid loss of volatile compounds and a mechanical stirrer, in order to control in a very accurate way the agitation rate. The reactor was put in a thermostatic oil bath to maintain constant the reaction temperature. The protocol followed for all the experiments was:
(a) charge of about 200 g of CPO;
(b) turning on the stirrer and the thermostatic bath to facilitate the palm oil dissolution;
(c) determination of the initial FFA weight by colorimetric titration, using phenolphthalein as indicator;
(d) addiction of the catalyst (14.1159 g in the initial run; all the following were performed without discharging it);
(e) after the achievement of the temperature set, addition of an amount of methanol to have a molar methanol–FFA ratio of 5, suitable for having only one liquid phase in the system.
This latter action corresponds to time zero.
A first set of 15 experiments were carried out at 60 °C and at an agitation of 250 rpm. The CPO used was characterized by an acid content of 3.50 wt%. The FFA conversion was evaluated by titrating a sample of about 1 mL of reaction mixture every hour. A second set of 16 batch experiments was performed increasing the initial FFA content by the addiction of oleic acid (Sigma Aldrich, purity >99%) with the aim to study the stability of carotenes at different FFA concentrations (from 3.50 wt% up to 7.70 wt%). The carotenes content was measured by monitoring the value of absorbance at 443 nm using a T60 (PG LTD) UV-vis spectrophotometer every two hours. The sample for the UV-vis analysis was prepared by diluting 1 mL of reaction sample 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 v/v with hexane.21 The catalyst used in the first experimental run was always reused for all the 31 runs to study the stability of the resin during the esterification reaction. Some of the results in terms of FFA abatement are shown in Fig. 1 as an example.
:70 v/v with hexane.21 The catalyst used in the first experimental run was always reused for all the 31 runs to study the stability of the resin during the esterification reaction. Some of the results in terms of FFA abatement are shown in Fig. 1 as an example.
 | ||
| Fig. 1 CPO deacidification results, FFA weight percentage versus time. | ||
The FFA content in oil decreases faster in the first and in the second experimental run due to the adsorption of water onto the resins. In fact, the catalyst was charged in its dry form the first time and the water produced by the reaction equilibrated onto the catalyst surface. After the water adsorption equilibrium was reached, the performances of the Amberlyst 46 resin remained stable and the final conversion was constant in all the further experiments.
The carotenes content in the CPO used for the experiments was 353 ppm. In Fig. 2 the final concentration of carotenes is reported at the end of 6 hours of reaction versus the number of the first set of experimental runs (performed with constant FFA initial concentration). This is lower than the initial value only in the first three runs, while for the others it reaches a plateau corresponding to the carotenes content of CPO. This observed decrease in carotenes concentration is due to the adsorption of these molecules on the catalyst particles, and, for this reason, it can be concluded that at the operative conditions chosen, i.e. 60 °C and room pressure, the concentration of the carotenes remains constant during the reaction.
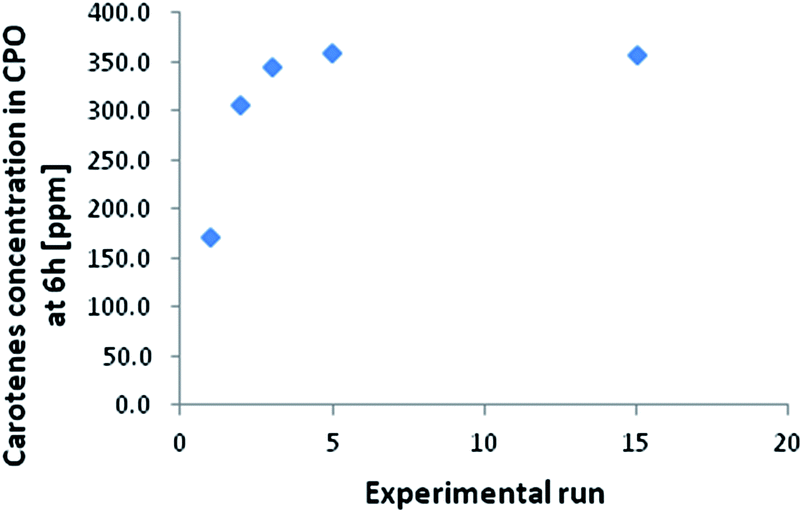 | ||
| Fig. 2 Carotenes concentration at the end of the experimental runs (6 h each). | ||
In order to deeper investigate the adsorption of carotenes on the catalyst surface, a sample of fresh resin and one of the same after all the experimental determination were observed using an optic microscope and the images are reported in Fig. 3.
 | ||
| Fig. 3 Amberlyst 46 images (×15) after and before all the experimental CPO deacidification tests. | ||
The magnifications show clearly that onto the resin surface there are species adsorbed. In a Soxhlet apparatus the total extraction of these products was carried out using n-hexane as solvent and then measuring the UV-vis spectra we noticed the presence of the typical carotenes absorption band. This result confirms that the carotenes are adsorbed on the resin, probably because of the chemical affinity between carotenes and the polymeric matrix of the catalyst.
The stability of the carotene adsorbed on the resin was evaluated by putting two samples of 2 g each of used catalyst in inert (N2) and oxidizing (O2) static atmosphere respectively. After 6 days an extraction using 15 mL of n-hexane was made and the absorbance for both the samples was registered. The results are shown in Fig. 4.
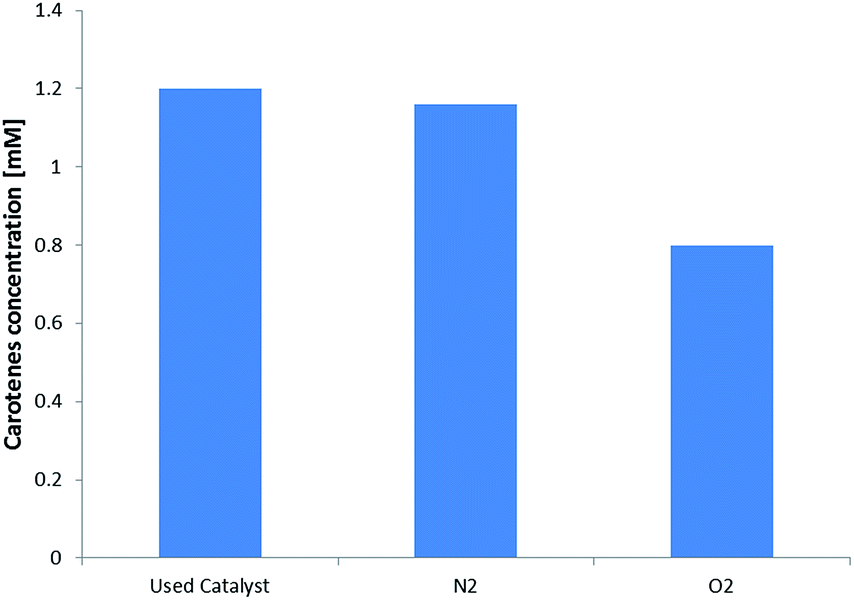 | ||
| Fig. 4 Carotenes concentration in 15 mL n-hexane extracts for 2 g of: (i) used catalyst after all the experimental runs; (ii) used catalyst in inert atmosphere (N2); and (iii) in oxidizing atmosphere (O2). | ||
The amount of carotenes remained stable after 6 days on the resin, meaning that the acid sites of the catalyst do not act on the degradation of carotenes. Differently, under oxygen atmosphere, the degradation of carotenes occurred and about the 40% of carotenes molecules were oxidized. Considering Henry et al.22 the time necessary to achieve this degradation value is about 9.4 days, however, considering that our experimental determination was made under an oxygen partial pressure of 1 atm, instead of about 0.2 atm, we can suppose that the adsorption on the resin surface limits the carotenes degradation. Further experimental verification are currently ongoing to correlate this decrease in oxidation rate to the polymeric matrix. Considering Fig. 2 and using the molecular weight of beta-carotene as representative for all the carotenes group, a total of 71 mg of carotenes were adsorbed onto the resin surface. This estimation was made considering that for every experimental run 200 g of CPO were used. From the first column of Fig. 4 it is possible to calculate the amount of carotenes captured by the catalyst, that is 68 mg of carotenes, confirming the mass balance of the collected experimental data.
The effect of FFA initial content on carotene preservation was studied in the second set of experimental runs, always with the same catalyst weighted at the beginning of this study. Fig. 5 shows both the final FFA conversion and the carotenes content in CPO (time = 6 h) versus the initial acidity of CPO.
 | ||
| Fig. 5 Experimental FFA conversion (blue points) and final carotenes content (red squares) versus initial FFA content in CPO. | ||
The results show that the concentration of carotenes remained stable and thus the presence of FFA does not influence the preservation of these important molecules. This result is noteworthy because the FFA content in an oil is not constant, but varies depending on the type of oil, the harvesting period and the manufacturing process.23 On the other hand, the final conversion of FFA tends to remain stable, showing again that the performances of the catalyst are not affected by the carotenes.
Conclusions
Carotenes content was preserved in the deacidification of CPO using an esterification reaction with methanol catalysed by an acid ion exchange resin (Amberlyst A46) at mild conditions of temperature and pressure. Moreover the adsorption of carotenes on the resin surface was observed and preliminary results suggest that this phenomenon contributes to make carotenes more stable to air oxidation. Different initial FFA content in CPO are not influent on carotene preservation. Satisfactory free fatty acid conversion was obtained and the catalyst performance was proved to be stable even after 186 hours of work.Notes and references
- R. Verwaal, J. Wang, J. P. L. Meijnen, H. Visser, G. Sandmann, J. A. Van den Berg and A. J. J. Van Ooyenl, Appl. Environ. Microbiol., 2007, 73(13), 4342 CrossRef CAS PubMed.
- H. T. Gordon and J. C. Bauernfeind, Crit. Rev. Food Sci. Nutr., 1982, 18(1), 59 CrossRef CAS PubMed.
- R. Davarnejad, K. M. Kassim, A. Zainal and S. A. Sata, J. Food Eng., 2008, 89, 472 CrossRef CAS PubMed.
- C. K. Ooi, Y. M. Choo, S. C. Yap and A. N. Ma, Elaeis, 1996, 8, 20 CAS.
- C. Pinto, L. L. N. Guariero, M. J. C. Rezende, N. M. Ribeiro, E. A. Torres, W. A. Lopes, P. A. P. Pereira and J. B. Andrade, J. Braz. Chem. Soc., 2005, 16B, 1313 CrossRef PubMed.
- M. Canakci and H. Sanli, J. Ind. Microbiol. Biotechnol., 2008, 35, 431 CrossRef CAS PubMed.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405 CrossRef CAS.
- M. K. Lam, K. T. Tan, K. T. Lee and A. R. Mohamed, Renewable Sustainable Energy Rev., 2009, 13, 1456 CrossRef CAS PubMed.
- R. Tesser, M. Di Serio, L. Casale, L. Sannino, M. Ledda and E. Santacesaria, Chem. Eng. J., 2010, 161, 212 CrossRef CAS PubMed.
- C. L. Bianchi, D. C. Boffito, C. Pirola, S. Vitali, G. Carvoli, D. Barnabè and A. Rispoli, Biodiesel feedstocks and processing technology, ed. M. Stoytcheva and G. Montero, Intech, 2011 Search PubMed.
- C. L. Bianchi, D. C. Boffito, C. Pirola and V. Ragaini, Catal. Lett., 2009, 134, 179 CrossRef.
- D. C. Boffito, C. Pirola and C. L. Bianchi, Chem. Today, 2012, 30, 42 CAS.
- M. J. Haas, P. J. Michalski, S. Runyon, A. Nunez and K. M. Scott, J. Am. Oil Chem. Soc., 2003, 80, 97 CrossRef CAS.
- C. Pirola, C. L. Bianchi, D. C. Boffito, G. Carvoli and V. Ragaini, Ind. Eng. Chem. Res., 2010, 49, 4601 CrossRef CAS.
- A. Chakrabarti and M. M. Sharma, React. Polym., 1993, 20, 1 CrossRef CAS.
- M. Jiménez, J. Santamaría-González, P. Maireles-Torres and A. Jiménez-López, Appl. Catal., B, 2011, 103(1–2), 91 CrossRef PubMed.
- L. Bernhard, M. E. Russbueldt and F. Wolfgang, Appl. Catal., A, 2009, 362, 47 CrossRef PubMed.
- N. Özbay, N. Oktar and A. Tapan, Fuel, 2008, 87(10–11), 1789 CrossRef PubMed.
- C. Pirola, F. Manenti, F. Galli, C. L. Bianchi, D. C. Boffito and M. Corbetta, Chemical Engineering Transactions, 2014, 37, 553 Search PubMed.
- C. Pirola, F. Galli, C. L. Bianchi, D. C. Boffito, A. Comazzi and F. Manenti, Energy Fuels, 2014, 28, 5236 CrossRef CAS.
- G. Britton, UV/visible spectroscopy, Carotenoids: Spectroscopy, ed. G. Britton, S. Liaaen-Jensen and H. Pfander, Birkhäuser, 1995, vol. 1B, pp. 13–62 Search PubMed.
- L. K. Henry, G. L. Catignani and S. J. Swartz, J. Am. Oil Chem. Soc., 1998, 75(7), 823 CrossRef CAS.
- R. D. O'Brien, Fats and Oils: Formulating and Processing for Applications, CRC Press, 2008, 3rd edn, Illustrations 33, p. 680 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |