
Functionalized single walled carbon nanotubes facilitate efficient differentiation of neuroblastoma cells in vitro
Abhinoy Kishorea,
Kaushiki Biswasa,
Vijaykameswara Rao Nb,
Raja Shunmugam*b and
Jayasri Das Sarma*a
aDepartment of Biological Sciences, Indian Institute of Science Education and Research-Kolkata (IISER-K), Mohanpur Campus, Mohanpur-741252, Nadia, West Bengal, India. E-mail: dassarmaj@iiserkol.ac.in
bPolymer Research Center, Department of Chemical Sciences, Indian Institute of Science Education and Research-Kolkata (IISER-K), Mohanpur Campus, Mohanpur-741252, Nadia, West Bengal, India. E-mail: sraja@iiserkol.ac.in
First published on 30th September 2014
Abstract
Single-walled carbon nanotubes (SWNTs) have been increasingly used as scaffolds for neuronal growth and differentiation. In this report, we have investigated how biocompatible functionalized SWNTs can affect the neuronal growth and morphology of an established neuronal cell line neuro2a (N2a). Interestingly, SWNTs covalently attached with biocompatible poly-D-lysine (PDL), facilitate neuronal growth and differentiation, neuronal growth on SWNTs with a combination of poly-ethylene glycol (PEG) with laminin (LA), a positive control molecule, is also studied. As expected, the mean length of the dendritic processes and axons is significantly larger with PDL as well as PEG–LA functionalized SWNTs compared to the control molecules. Most importantly, N2a cells retain neuronal morphology in the culture for a longer time (21 days) in presence of SWNT–PDL or SWNT–PEG LA, compared to the control culture. SWNTs modified neuronal cells in culture are infected with enhanced green fluorescent protein (EGFP) tagged recombinant neurotropic mouse hepatitis virus (MHV) to demonstrate that SWNT modulated N2a cells are a promising substrate for understanding the viral antigen spread and persistence as a proof of hypothesis.
Introduction
Neuro2a (N2a) is a mouse neuroblastoma cell line that has been extensively used to study neuronal differentiation, axonal growth and signalling pathways. A convenient characteristic of these cells is their ability to differentiate into neurons within a few days. The growth and differentiation of neural stem cells on nanotube substrates has recently been demonstrated.1 Recently, in one of our studies, N2a cells are used to understand the mechanisms of the spread of neurotropic mouse hepatitis virus RSA59 and its persistence. RSA59 is one of the EGFP expressing isogenic recombinant strains of mouse hepatitis virus MHV-A59.2–4 MHV-A59 or its isogenic spike protein recombinant strain RSA59 causes viral meningitis, encephalitis (neuroinflammation) and demyelination (loss of myelin sheath) which mimics certain pathology of human demyelinating disease multiple sclerosis (MS).2,5 Myelin forms an insulating sheath surrounding axons in the central and peripheral nervous systems and is essential for rapid action potential propagation. Demyelination is believed to be an acquired immune mediated disorder in which normally formed myelin degenerates, exposing axons to the extracellular environment.6–8 The result is dysfunction and altered regulation of normal neuron-to-neuron communication and in many cases, varying degrees of axonal degeneration. Although demyelination is the major manifestation of most of the demyelinating diseases, recent studies have clearly documented concurrent axonal loss to varying degrees resulting in long-term disability.9 Axonal injury may be secondary to myelin damage (outside-in model) or myelin damage occurring secondary to axonal injury (inside-out model). Mouse hepatitis virus induced neuroinflammation is an excellent animal model to understand the mechanism of axonal loss and demyelination.10 Based on previous experimental evidences, it can also be argued that the spread of viral antigen from neuron to neuron and from neuron to glial cells plays a critical role in the induction of chronic stage demyelination.3,5 Moreover, it has been demonstrated that axonal transport of viral particles is an important mechanism mediating not only the extent of axonal damage but also the subsequent induction of demyelination in the spinal cord.11 But the underlying mechanism of neuronal transport of neurotropic MHV strains is not known due to the complex in vivo cellular network of the central nervous system (CNS). To avoid such complex nature of the in vivo system various studies of viral spread is conducted in vitro with primary hippocampal neuronal culture and dorsal root ganglionic neuronal culture.5,12 Though primary culture can mimic the in vivo system very closely, its tedious, cost-effective and time consuming nature makes this system inappropriate for long term viral spread and persistence studies. Alternatively, neuroblastoma cells could be used as a cell culture model to study viral spread and persistence. However, the majority of N2a cells retain its embryonic nature (mother glial which is positive for GFAP; glial fibrillary acidic protein) which makes the culture system heterogeneous and non-compatible for in vitro studies. There are a number of reports to evaluate the effect of a number of growth factors and hormones to induce neuronal differentiation of N2a cells to the homogeneous neuronal phenotype.13 In this study single walled carbon nanotubes (SWNTs) are functionalized with poly-D-lysine (PDL) or with polyethylene glycol (PEG) or PEG in combination with laminin (LA) as a substrate/scaffold to modulate the N2a cells into a more neuronal phenotype that are used for in vitro viral spread and viral persistence studies.Carbon nanotubes (CNTs), known for their size and shape similar to neuronal processes, and strong, flexible, conductive nature, were considered as substrates for neuronal growth and development for a long time.14,15 But recently, it has been observed that the growth of the neuron could be improved by modifying CNTs with either a biologically active chemical compound (4-hydroxynonenal; by physisorption)15,16 or by modifying the charge of the CNT substrate with chemical modifications. Functionalization with carboxyl groups or poly-m-aminobenzene sulfonic acid or ethylenediamine create negatively, zwitterionic or positively charged nanotubes, respectively.17 It has also been observed that if the substrate is positively charged, attachment and outgrowth of the neurite is much more pronounced compared to either zwitterionic or negatively charged nanotubes. Previously thin films of layer-by-layer assembled composites of SWNT coated with laminin have been shown to induce differentiation of neural stem cells and serve as a potential foundation material for neural electrodes.18 In corroboration, the current study further investigates the role of SWNT covalently linked to PDL or LA. The chemical modification and bio-functionalization with PDL or PEG–LA has resolved the non-solubility issue of SWNTs in aqueous media and made them suitable to generate a new class of bioactive carbon nanotubes which neither induce impurities nor stimulate any biological activity in the cells. In this report, we have clearly demonstrated that neither SWNT nor PDL or LA or PEG or PEG–SWNT alone could promote any additional neuronal differentiation with stable morphology for nearly 21 days in comparison to the control untreated cells. To the best of our knowledge, this is the first report which convincingly shows that the functionalized SWNTs are better inducers for neuronal differentiation.
Experimental
Materials
Poly-D-lysine hydrobromide (PDL) [Mw = 512; Ln = 4 by MALDI],19 SWNT (single-walled carboxylic acid functionalized carbon nanotubes), DCC (N,N′-dicyclohexylcarbodiimide), PEG (polyethylene glycol), PDL (poly-D-lysine), LA (laminin) and DMF (dimethyl formamide) were purchased form Sigma-Aldrich, USA. DMF was dried and used for the reactions.Functionalization of carbon nanotubes with PDL
Ten milligrams (10 mg) of the SWNT carboxylic acid was added to either 60 mg of PDL or 60 mg of polyethylene glycol (PEG), and 20 mg of DCC were added to each completely dried 4 neck flask. 60 mL of dry DMF was added as solvent in the PDL reaction tube and 10 mL of DMF was added to PEG–LA reaction tube and both reaction mixtures were stirred at 50 °C for 24 h under nitrogen flow. After that, the SWNT–PDL or SWNT–PEG–LA assembly was collected using a polycarbonate membrane with 200 nm pores. Finally, the product was thoroughly washed with alcohol and water, and dried under vacuum, to remove any unbound PDL or PEG–LA and DCC (Scheme 1). Formation of the product was confirmed by FT-IR spectroscopy. FT-IR spectra were obtained on a FT-IR Perkin-Elmer spectrometer at a nominal resolution of 2 cm−1. Low resolution transmission electron microscopy (TEM) was performed on a JEOL 200 CX microscope. TEM grids were purchased from Ted Pella, Inc. and consisted of 3–4 nm amorphous carbon film supported on a 400-mesh copper grid.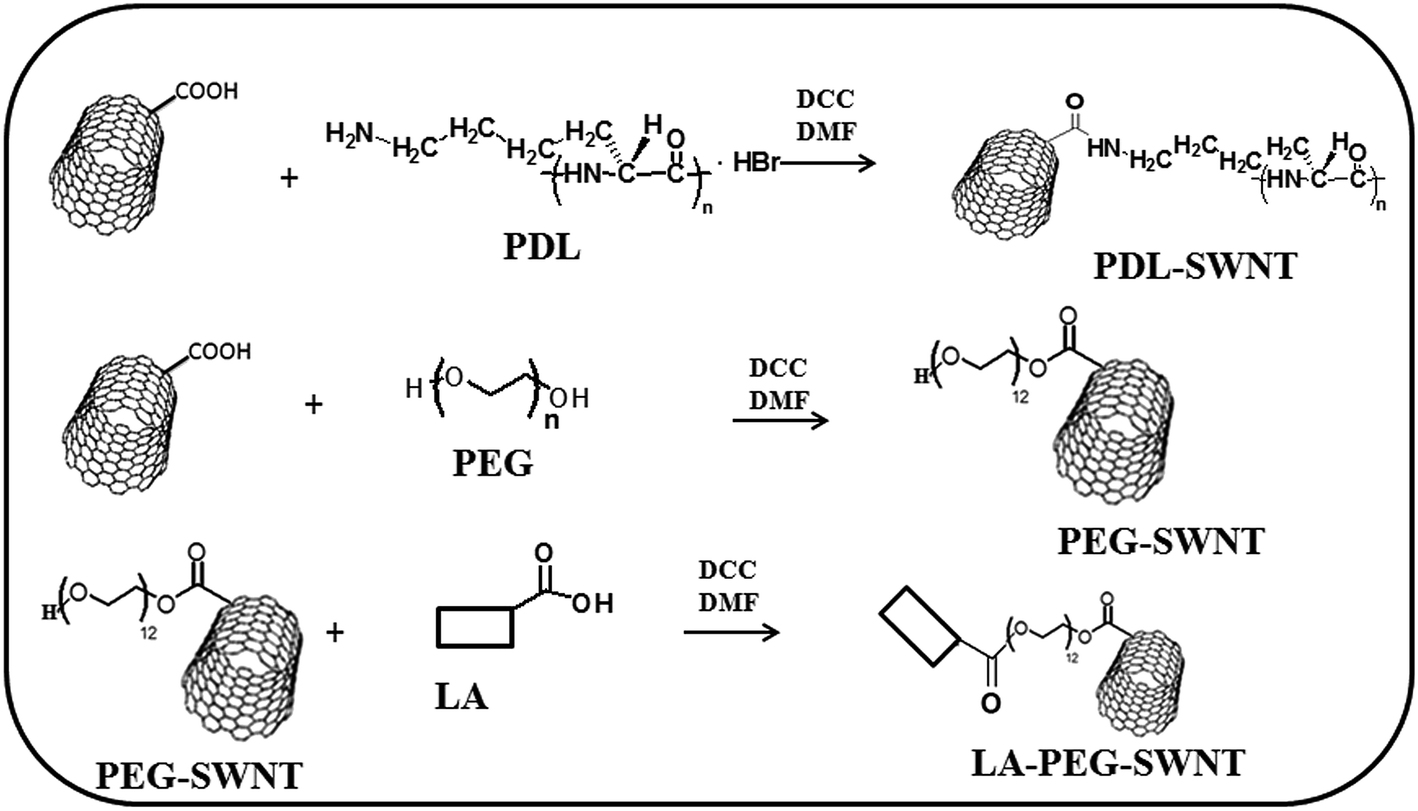 | ||
| Scheme 1 Schematic representation of the synthesis of PDL, PEG and PLA attached to SWNT. | ||
N2a cells
N2a is a mouse neuroblastoma cell line (CCL-131) obtained from American cell type culture collection. Briefly, N2a cells were maintained in MEM with 10% FBS (Gibco) and penicillin (10.000 U mL−1) and streptomycin (100 μg mL−1) (HyClone) in presence of 5% CO2 at 37 °C in between passage 2–5 and were seeded onto either 24 well tissue culture plate (Nunc) or etched coverslips (Belco) coated with 0.01 mg mL−1 poly-D-lysine hydrobromide.Treatment of N2a cells with different functionalized SWNTs
Functionalized SWNT–PDL (50 μg mL−1), SWNT–PEG (50 μg mL−1), SWNT–PEG–LA (50 μg mL−1), and only PDL (50 μg mL−1), LA (250 μg mL−1), PEG (250 μg mL−1) and SWNT (50 μg mL−1) were suspended in cell culture medium (MEM; as specified in N2a cell medium) by using a bath sonicator for 1 h and added onto 80% confluent monolayers of N2a cells. Mock (PBS) treated culture was used as a control. After 72 h of treatment cells were immunostained with anti-neurofilament antibodies in combination with Texas red conjugated goat anti-mouse IgG as secondary antibodies. Fluorescence images were acquired with a Hamamatsu Orca-1 CCD camera and were analyzed by Image ProPLus image analysis software (Media Cybernetics, Silver Spring, MD) and ImageJ software.Treatment of N2a cells with FUDR before treating with functionalized SWNTs
Functionalized and characterized SWNT–PDL (50 μg mL−1) was suspended in cell culture medium (MEM; as specified in N2a cell medium) by using a bath sonicator for 1 h. N2a cells were placed on PDL coated coverslips and maintained until 80% confluency. Non neuronal cells were eliminated from the confluent monolayer cells by adding a mixture of fluorodeoxyuridine and uridine containing medium (FUDR) (10 μM). After 24 h fluorodeoxyuridine and uridine containing medium was replaced with either fresh N2a specified medium or medium with 25, 50, 100 and 200 μg of suspended SWNT–PDL and cells were incubated for 96 h. Similarly, parallel control cultures were maintained in fresh medium without FUDR and SWNT–PDL treatment for 48 h after 80% confluency to maintain the proper time frame of neuronal growth in culture.Characterization of neuronal phenotype
Neurons from SWNT–PDL treated culture as well as control cultures were identified based on their morphological features using differential interference contrast (DIC) and fluorescence microscopy. This method of identification had previously been confirmed as reliable by labelling for immunofluorescence with primary antibodies directed against axonal microtubule marker TAU,20 dendritic microtubule marker MAP2 (2a+2b),21 neurofilament marker (NFM)22 and glial fibrillary acidic protein (GFAP) astrocyte specific marker. Detailed specification of primary and secondary antibodies and their working dilutions were mentioned in Table 1. Briefly, N2a cells were fixed with 5% ice cold glacial acetic acid in absolute alcohol for 10 min, washed with PBS (with Ca2+ and Mg2+) and permeabilized with PBS–TX (PBS with 0.4% Triton-X) followed by blocking in PBS–TX–GS (PBS–TX with 2.5% normal goat serum). After blocking, cells were incubated with monoclonal primary antibodies directed against TAU, NFM, MAP2, and GFAP diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in PBS–TX–GS and incubated for 2 h at room temperature. Cells were then successively washed with PBS (with Ca++ and Mg++) three times with 5 min of incubation each time and incubated with Texas red conjugated anti-mouse secondary antibody (Jackson Immunoresearch) for 1 h at room temperature. Cells were again washed three times with PBS (with Ca++ and Mg++), followed by a PBS only wash for 5 min, mounted with Vectashield (Vector Laboratories) and visualized by fluorescence microscopy using an Olympus IX-81 microscope system with a 40× UPlanApo objective (1.0 numerical aperture). Images were acquired with a Hamamatsu Orca-1 CCD camera and analyzed by Image ProPLus image analysis software (Media Cybernetics, Silver Spring, MD). Parallel DIC images were acquired for each fluorescence image to observe the neuronal phenotype in detail. Photo bleaching, if there was any, was controlled by inserting neutral density filters and an electronic shutter.
:100 in PBS–TX–GS and incubated for 2 h at room temperature. Cells were then successively washed with PBS (with Ca++ and Mg++) three times with 5 min of incubation each time and incubated with Texas red conjugated anti-mouse secondary antibody (Jackson Immunoresearch) for 1 h at room temperature. Cells were again washed three times with PBS (with Ca++ and Mg++), followed by a PBS only wash for 5 min, mounted with Vectashield (Vector Laboratories) and visualized by fluorescence microscopy using an Olympus IX-81 microscope system with a 40× UPlanApo objective (1.0 numerical aperture). Images were acquired with a Hamamatsu Orca-1 CCD camera and analyzed by Image ProPLus image analysis software (Media Cybernetics, Silver Spring, MD). Parallel DIC images were acquired for each fluorescence image to observe the neuronal phenotype in detail. Photo bleaching, if there was any, was controlled by inserting neutral density filters and an electronic shutter.
| Primary antibodies (dilutions 1:100) |
Cell specific marker | Secondary antibodies (dilutions 1:100) |
|---|---|---|
| Mouse monoclonal anti-TAU, Sigma | Axons and cell soma | Texas red conjugated goat anti-mouse IgG Jackson Immunoresearch |
| Mouse monoclonal anti-MAP2 (2a+2b), Sigma | Dendrites and dendritic processes | Texas red conjugated goat anti-mouse IgG, Jackson Immunoresearch |
| Mouse monoclonal anti-Neurofilament (NFM), Sigma | Axons and cell soma | Texas red conjugated goat anti-mouse IgG, Jackson Immunoresearch |
| Mouse monoclonal anti-GFAP, Sigma | Astrocytes and mother glia | FITC-conjugated goat anti-mouse IgG, Jackson Immunoresearch |
Infection of N2a cells with neurotropic mouse hepatitis virus
A 80% confluent culture of N2a cells grown on PDL coated glass coverslips were infected with RSA59, a neurotropic recombinant mouse hepatitis virus (MHV) strain tagged with enhanced green fluorescent protein (EGFP),4 at MOI of 1:1. Viral particles were allowed to adsorb for 1 h. Viral inoculum was then washed with PBS and replaced with fresh medium (MEM + 10% FBS + 1% Penstrep). At 0, 6, 12 and 20 h post infection (p.i.), cultures were monitored for replication and viral antigen (EGFP) spread by Olympus IX81 fluorescence microscopy using a suitable filter.
Infection of SWNT–PDL treated N2a cells with RSA59
RSA59 is widely used in the study of viral tropism in different neural cell types and neural cell to cell spread. The modified SWNT–PDL treated culture created in this study could, indeed, be used to study the viral spread and persistence. FUDR treated N2a cells maintained in the presence of SWNT–PDL onto PDL coated glass coverslips were infected at a multiplicity of infection (MOI) of 1:1 (1 viral particle for one cell) with RSA59 or mock infected with PBS. After allowing viral adsorption for 1 h as described above, cells were washed and placed in fresh media without virus. Viral replication and viral antigen spread was monitored by EGFP fluorescence for 24, 48 and 72 h. For neurons that extended processes beyond the field of view, multiple images were acquired and merged together using adobe Photoshop CS5 (Adobe system Inc., San Jose, CA). Neuronal morphological characteristics were quantified using the ImageJ program.
Results and discussion
Single-walled carbon nanotubes (SWNTs) functionalized with carboxylic acid groups were covalently attached to biocompatible PDL or PEG–LA, by using the coupling reagent DCC in DMF at 50 °C under nitrogen flow. Formation of the product was confirmed by FT-IR spectroscopy. FT-IR spectra were obtained on a FT-IR Perkin-Elmer spectrometer at a nominal resolution of 2 cm−1. The FTIR spectra of poly-D-lysine, SWNT, and PDL attached SWNT are shown in Fig. 1a–c. From Fig. 1a it was observed that the vibration of amide I appeared at 1620 cm−1, that of amide II appeared at 1536 cm−1, and that of free amino groups (NH3+) appeared at 3300 cm−1. The stretching frequency at 1730 cm−1 was due to the carboxylic acid of SWNTs (Fig. 1b). The covalent attachment of PDL to SWNT was confirmed by (Fig. 1c) the shifting of carboxylic acid stretching mode from 1730 cm−1 to 1654 cm−1, due to the formation of a new amide bond between SWNT and PDL. Single-walled carbon nanotubes functionalized with carboxylic acid groups (SWNTs) were covalently attached to biocompatible polyethylene glycol (OH–PEG–OH), by using the coupling reagent DCC in DMF at 50 °C under nitrogen flow. Formation of the product was confirmed by FT-IR spectroscopy (Fig. 2a–c). In the FT-IR spectrum of SWNT–PEG, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of a carboxyl group was shifted from 1642 to 1628 cm−1, which indicated the formation of product as shown in the Fig. 2b. To get SWNT–PEG, the hydroxyl group was reacted with the carboxylic acid group of LA, by using coupling reagent DCC in DMF at 50 °C under nitrogen flow. In the FT-IR spectrum of SWNT–PEG–LA, the CO stretching band of a carboxyl group was shifted from 1670 cm−1, and disappearance of 1628 cm−1, indicated the formation of product as shown in the Fig. 2c. The Transmission Electron Microscopy (TEM) analysis of PDL functionalized SWNTs also supported the attachment of PDL on to the SWNT (Fig. 3a–c). The selective area electron diffraction (SAED) patterns observed from the TEM (Fig. 3d) suggested that those crystalline patterns were due to PDL.
O stretching band of a carboxyl group was shifted from 1642 to 1628 cm−1, which indicated the formation of product as shown in the Fig. 2b. To get SWNT–PEG, the hydroxyl group was reacted with the carboxylic acid group of LA, by using coupling reagent DCC in DMF at 50 °C under nitrogen flow. In the FT-IR spectrum of SWNT–PEG–LA, the CO stretching band of a carboxyl group was shifted from 1670 cm−1, and disappearance of 1628 cm−1, indicated the formation of product as shown in the Fig. 2c. The Transmission Electron Microscopy (TEM) analysis of PDL functionalized SWNTs also supported the attachment of PDL on to the SWNT (Fig. 3a–c). The selective area electron diffraction (SAED) patterns observed from the TEM (Fig. 3d) suggested that those crystalline patterns were due to PDL.
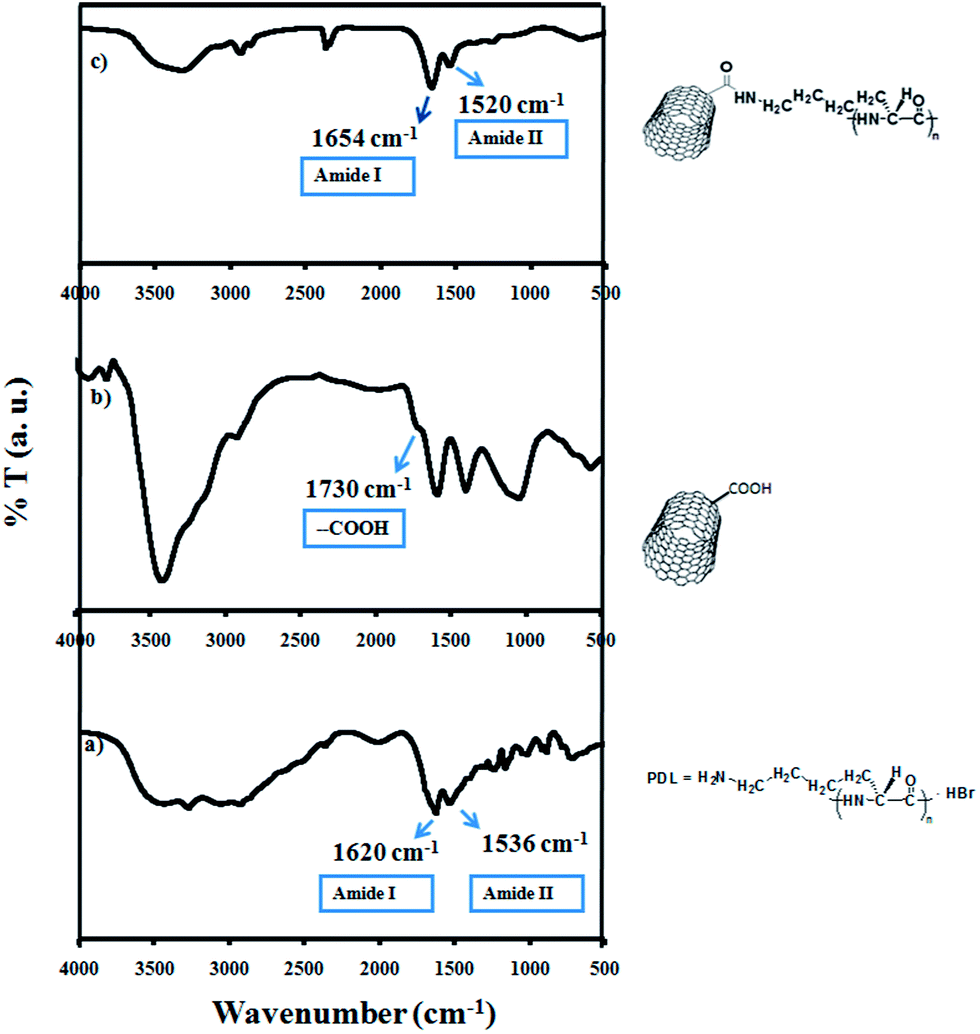 | ||
| Fig. 1 Infra red spectra of (a) PDL, (b) SWNT and (c) PDL functionalized SWNT. | ||
 | ||
| Fig. 2 Infra red spectra of (a) SWNT–PEG–LA, (b) SWNT–PEG and (c) PEG–OH. | ||
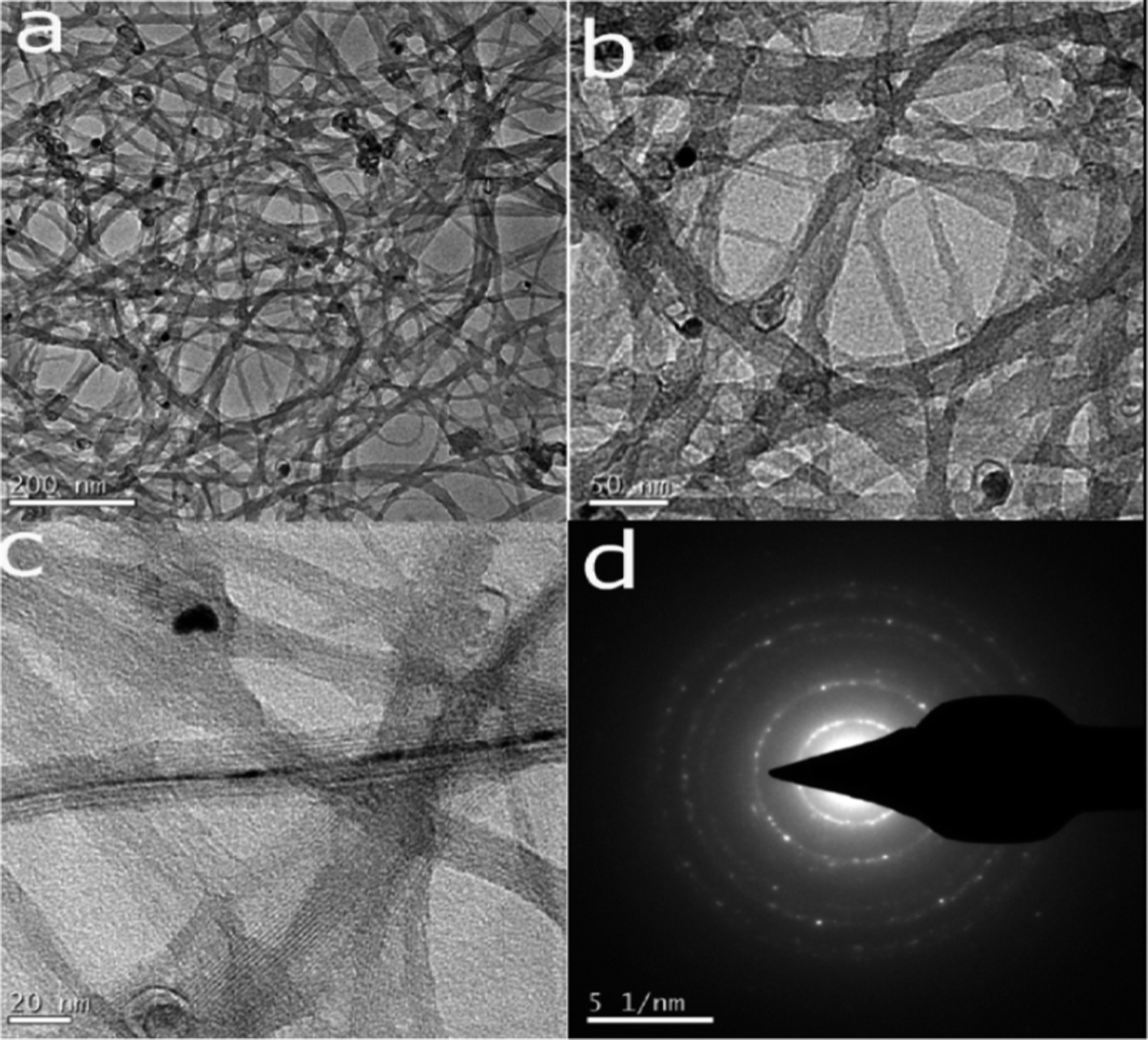 | ||
| Fig. 3 Transmission Electron Microscopy (TEM) images of PDL functionalized SWNT. Analysis of PDL functionalized SWNTs by TEM also supported the attachment of PDL on to the SWNT (a–c); stained with osmium tetroxide. The selected area electron diffraction pattern (SAED) suggested that those crystalline patterns were responsible for the PDL (d). | ||
Functionalized SWNT–PDL (50 μg mL−1), SWNT–PEG (50 μg mL−1), SWNT–PEG–LA (50 μg mL−1), along with free PDL (50 μg mL−1), LA (250 μg mL−1), PEG (250 μg mL−1) and SWNT (50 μg mL−1) were suspended in cell culture medium (MEM; as specified in N2a cell medium) by using a bath sonicator for 1 h and added onto 80% confluent monolayers of N2a cells plated onto a 24 well plate treated for cell culture. Mock (PBS) treated culture was used as a control. After 72 h post treatment, treated and control cells were observed in phase contrast microscopy for the neuronal phenotype (data not shown) and processed of neurofilament immunostaining for phenotypic characterization. Neurons from SWNT–PDL treated culture as well as control cultures were identified based on their morphological features using differential interference contrast (DIC) (data not shown) and neurofilament staining under fluorescence microscopy (Fig. 4). Neurofilament staining results demonstrated that SWNT either functionalized with PDL (Fig. 4F) or with PEG–LA (Fig. 4H) promoted differentiation of N2a cells into a more neuronal phenotype. Also, both dendritic processes and axonal projections were clearly defined as well as prominent when compared to the control N2a cells (non treated). In contrast no obvious phenotypic differentiation was observed between PDL (Fig. 4E) or LA (Fig. 4G) or SWNT–PEG (Fig. 4D), or PEG (Fig. 4C) or SWNT (Fig. 4B) treated N2a cells in comparison to control (non-treated) (Fig. 4A). No obvious cell toxicity was observed in any of the treatment conditions as checked by MTT assay (data not shown). But a large population of cells either treated with PDL alone or PEG or SWNT–PEG or untreated retained their embryonic features as cells in confluent culture and was largely positive for GFAP, which indicated that neuroblastoma cells in the mentioned culture condition did not completely differentiate into the neuronal phenotype (data not shown). PDL and LA modified SWNT provided comparable differentiation of neurons in culture, and so as PDL was less expensive compare to LA, PDL functionalized SWNT was used as scaffold for the rest of the studies.
 | ||
| Fig. 4 Differential neuron phenotype of N2a cells in presence or absence of functionalized SWNTs. PDL (F) or PEG–LA (H) modified SWNT treated N2a compared to control untreated (A) cells showed the maximum differentiation into dendritic processes and axonal projections with prominent synapse formations in culture with SWNT–PDL (shown by arrows). Neither SWNT (B), nor PDL (E) or LA (G) or PEG (C) or PEG–SWNT (D) alone could promote any additional neuronal differentiation compared to control untreated cells. | ||
N2a cells were plated onto PDL coated coverslips on 24 well plates. Non neuronal cells were eliminated from the confluent monolayer cells by adding a mixture of fluorodeoxyuridine and uridine containing medium (FUDR) (10 μM). After 24 h fluorodeoxyuridine and uridine containing medium was replaced with either fresh N2a specified medium or medium with 25, 50, 100 and 200 μg of suspended SWNT–PDL and cells were incubated for 96 h. Similarly, parallel control cultures were maintained in fresh medium without FUDR and SWNT–PDL treatment for 48 h after 80% confluency to maintain the proper time frame of neuronal growth in the culture. N2a cells treated with FUDR alone (without SWNT–PDL), died within 48 h. Indeed, at lower concentrations (less than 25 μg as studied) of SWNT–PDL treatment, N2a cells after FUDR treatment were observed to die within 48 h of treatment. The number of GFAP positive cells was drastically reduced (data not shown). But FUDR treated cells in the presence of SWNT–PDL (50 μg) maintained their neuronal phenotype until 10 days as tested. FUDR treatment helped to reduce the number of dividing cells and the majority of the post mitotically non-dividing cells were differentiated into neuronal phenotypes in the presence of SWNT–PDL.
Neurons from SWNT–PDL treated culture as well as control cultures were identified based on their morphological features using differential interference contrast (DIC) (Fig. 5A and B) and fluorescence microscopy. This method of identification had previously been confirmed as reliable by labelling for immunofluorescence with primary antibodies directed against axonal microtubule marker TAU (Fig. 5C and D), dendritic microtubule marker MAP2 (2a+2b) (Fig. 5E and F),21 neurofilament marker (NFM) (Fig. 5G and H) with TRITC conjugated anti-mouse secondary antibody and glial fibrillary acidic protein (GFAP) astrocyte specific marker (Fig. 5I and J) with FITC conjugated secondary antibody. Parallel DIC images and fluorescence images were acquired to observe neuronal phenotype in detail (Fig. 5). Observations by DIC microscopy revealed that the total number of processes, neurites originating from the cell body, for each neuron remained the same but the total outgrowth, the summed length of all processes and their branches, of each neuron was significantly greater in SWNT–PDL treated culture (Fig. 5B) compared to control (Fig. 5A). Immunophenotyping with different compartment specific antibodies in combination with DIC image analysis revealed that both SWNT–PDL treated and non treated cells expressed Tau (axon specific), but Map2 (2a+2b) (dendritic processes specific) expression was much less in non treated cells compared to the SWNT–PDL treated cells. The length of the dendritic processes with a higher level of expression of these compartment specific proteins may signify that SWNT–PDL and SWNT–PEG–LA helps in the differentiation process along with the growth.
 | ||
| Fig. 5 Morphological characterization of N2a cells with different neuronal compartment specific markers before and after treatment with SWNT–PDL. N2a cells were treated with FUDR and then with SWNT–PDL as described earlier in the manuscript. Neurons from SWNT–PDL treated culture as well as control cultures were identified based on their morphological features using differential interference contrast (DIC) microscopy (A and B), and fluorescence labelling of different neuronal compartment specific antibodies (C–H). As described in Table 1 Anti-Tau (red), Anti-MAP2 (red), Anti-Neurofilament (NFM) 200 (red), Anti-GFAP (green) antibodies are used to label axonal microtubule (C and D), dendritic microtubule (E and F), neurofilament protein (G and H) and glial fibrillary acidic protein (GFAP) (I and J) respectively and mounted with vectashield (Vector laboratories) with DAPI (blue, counterstained for nucleus). All images were captured in 40× objective and background staining was neutralized by using neutral density filter. Representative fields are shown in this figure. | ||
In the presence of SWNT–PDL or SWNT–PEG–LA, N2a cells acquired the differentiated neuronal phenotype and formed neural network with proper synapse formation, which was not evident in non treated N2a cells by anti-synaptophysin antibody (data not shown). Synapse formation was very much evident in SWNT–PDL treated cells (Fig. 4F; white arrows mark the synaptic junctions) and also confirmed by immunofluorescence with anti-synaptophysin antibodies (data not shown).
The mean neurite lengths had increased because the length of each process had increased (Fig. 6) though the number of processes remained the same. Images were taken for days 1, 2, 3, 4, 5, 6, 7 and day 20 from FUDR treated N2a cells grown in the presence and absence of SWNT–PDL. FUDR treated N2a cells in the absence of SWNT–PDL were found to be dead within 24 to 48 h of observation. In presence of SWNT–PDL, N2a cells were found to maintain their differentiated stages for 20 days. For measuring the neurite length ImageJ software was used and it was strictly noted that only isolated single neurons which did not form synaptic connections with other neurons were taken into consideration for the measurement. It was found that there was an increase in the area of the neuronal cell body in the DIC image (data is not produced). It was also found that neurites extended their maximum length until day 7 and attained their maximum differentiation on day 7 when treated with SWNT PDL. Thereafter, no further significant neurite growth was observed but it was also found that they retained their neuronal phenotype, until day 20.
 | ||
| Fig. 6 Average length of the neurite in SWNT–PDL treated N2a cell culture at different days post treatment. For neurite length measurements, SWNT–PDL treated and untreated control (minus SWNT–PDL) cultures were used and only isolated single neurons that did not form synaptic connections with other neurons were taken into consideration for measurement. Mean length of neurites were plotted against days of post treatment. Significant differences in length were observed for different days of SWNT–PDL treated cultures. Two tailed t-test were conducted in graph pad prism software and the p values were plotted as stars (*). **** (p value < 0.0001), *** (p value < 0.0003), ** (p value < 0.004). | ||
Mouse hepatitis virus is one of the neurotropic experimental mouse viruses which upon intra cranial (i.c.) inoculation can follow the axonal route to travel from brain to different anatomic regions of the CNS. Recent studies have demonstrated clear evidence of retrograde viral transport in an in vivo MHV model of optic neuritis23 and anterograde axonal transport in the myelitis and spinal cord demyelination model of MHV.5 The mechanism of viral axonal transport is not very clearly understood in vivo due to the complicated nature of different nerve tracks in the CNS and study of this mechanism demands a simple neuronal culture system which will support neurotropic mouse hepatitis virus infection spread and persistence.
To confirm whether N2a cells can support neurotropic MHV infection, an 80% confluent monolayer of N2a cells grown in the presence or absence of PDL–SWNT for 24 h were infected with RSA59 at a MOI of 1 or mock infected. At 12 h p.i. several infected cells were observed with very little spread in RSA59 infected cultures (data now shown), whereas by 20 h p.i., the number of EGFP-positive cells were significantly increased and RSA59 infection was observed to spread from one neuron to the next (Fig. 7), as nearly all cells in the culture contained viral antigen. At an earlier time post infection, (6 and 12 h p.i.) only a small percentage of cells were infected. This data is consistent with previous data on primary hippocampal neuron.5,12,24 As expected, very few discrete autofluorescent cells were observed in mock infected cultures (data not shown) at different time points post infection. N2a cells could be an appropriate model to study the viral replication and viral antigen spread in vitro but the embryonic nature of N2a cells is one of the major concerns for the viral antigen spread and persistence studies in vitro. Moreover, after 24 h most of the infected cells are dying either by forming syncytia or by the lytic nature of the cells which are not allowing the tracing of transneuronal viral spread and persistence in the neuron for longer periods of time.
 | ||
| Fig. 7 Infection of N2a cells with neurotropic Mouse Hepatitis Virus (MHV). Three day old confluent culture of N2a cells were infected with neurotropic recombinant mouse hepatitis virus, RSA59 tagged with EGFP at M.O.I of 2. After 20 h p.i. cells were washed with PBS (with Ca2+ and Mg2+) and mounted in Vectashield (Vector Laboratories). Fluorescence images and corresponding DIC images were captured on an Olympus IX 81 epifluorescent microscope. All through the studies a 40× objective was used to attain better resolution and a larger field. One of the representative fields containing 60–70% infected cells is shown in this figure (A) and corresponding DIC image (B) (scale bar; 50 μm length). The thicker arrow indicates syncytia formation and the thinner arrow shows the neuronal processes. | ||
The modified SWNT–PDL treated culture created in this study could, indeed, be used to study the viral spread and persistence. FUDR treated N2a cells maintained in presence of SWNT–PDL on PDL coated glass coverslips were infected at a multiplicity of infection (MOI) of 1:1 (1 viral particle for one cell) with RSA59 as described above, and monitored for replication and viral antigen (EGFP) spread by fluorescence microscopy (Fig. 8) for 7 days. At 24, 48 and 72 h post infection, cells were fixed by ice cold absolute ethanol for 10 min and processed for imaging. For neurons that extended processes beyond the field of view, multiple images were acquired and merged together using adobe Photoshop CS5 (Adobe system Inc., San Jose, CA). Neuronal morphological characteristics were quantified using the Image program. Differentiated N2a cells in SWNT–PDL treated cultures when infected with EGFP tagged RSA59 (neurotropic strain of MHV-A59) at 1 M.O.I. retain the EGFP fluorescence both in the cell body as well as in dendrites and axons, which indicated that viral particles were replicating in the cell body and could spread along the dendritic processes (retrograde fashion) as well as through axonal processes (anterograde fashion) as shown in vivo.
 | ||
| Fig. 8 RSA59 infection persists for 7 days in SWNT–PDL (SWNT–PDL) treated differentiated N2a cells. SWNT–PDL treated differentiated cells were maintained in the culture for 7–10 days and then infected with 1 M.O.I of RSA59. RSA59 viral particles were allowed to adhere to the cells for 1 h and then incubated with N2a medium in the presence of SWNT–PDL for 7 days. At every 24 h interval, infected cells were observed under the fluorescence microscope for viral antigen spread. At different time intervals, p.i. infected cells were mounted for microscopy and observed for persistent viral infection. Three representative images from three different time points p.i.; 24 h p.i. (A) 48 h p.i. (C) and 72 h p.i. (E) were shown with corresponding differential interference contrast (DIC) images (B, D and F). In most of the observed microscopic fields after 48 h, SWNT–PDL treated neurons extended processes beyond the field of view, so multiple images were acquired and merged together using adobe Photoshop CS5. Scale bar = 50 micron. | ||
Laminin is a matrix protein that has been extensively used to culture primary neuronal cells and neuronal cell lines and has been found to be an excellent cell adhesion molecule and a potent stimulator of neurite outgrowth and axonal regeneration.25–29 The cationic nature and unique adhesive property of PDL with biomolecules of cell structure and biocompatibility with cell behaviour simply enhances its adhesiveness with plastic and glass surfaces for many anchorage-dependent cells and specifically primary cell adhesion to the plates.30 Recent studies demonstrated that covalent binding of PDL to metallic chips enhance neurite outgrowth and neurite length.31 Combining the cell binding prospects of PDL and LA along with the fascinating physical properties of nano materials, this work investigates functionalization of CNTs with PDL or LA. The chemical modification and bio-functionalization with PDL or PEG–LA resolved the non-solubility issue of CNTs in aqueous media and made CNTs suitable to generate a new class of bioactive carbon nanotubes which neither induce impurities nor stimulate any biological activity in the cells. Although PDL and LA is important for the growth of the N2a cells, the conductivity of SWNT supports the growth and differentiation of the neuronal cells and inhibits gliogenesis. There is precedence that conductive substances are advantageous for use as neuronal implants in vivo, supporting neuronal growth and exhibiting less gliosis than nonconductive substances like Teflon implants when inserted into rat cortex.32
The PDL functionalized SWNT treated persistent N2a cell culture system could be an appropriate model to study the molecular mechanisms of kinesins (anterograde) and dyneins (retrograde) mediated axonal transport and fusion of the viral particle at the axon end or dendritic spine and shafts. Insights from this viral axonal transport studies will help to shed some light to understand the cellular and molecular mechanisms of axonal transport of other neurotropic viruses like herpes simplex virus and pseudorabies virus which are also known to follow axonal transport as the conduit to reach the neuronal cell body.33,34
Conclusions
Functionalized SWNT with biocompatible PDL in suspension modulates neuronal growth. Most of the studies on functionalized SWNT were concentrated to study how the modified SWNT qualities play a role in the process of growth cone motility, neurite branching and differentiation in adherent culture. But this is the first time we have demonstrated that the application of functionalized SWNT with PDL in suspension can modulate neuronal growth and differentiation. Moreover, this is the first demonstration of the application of functionalized biocompatible SWNT in the field of neurovirology. In the future this study will be extended into functionalization of SWNT with other bioactive compounds like poly-l-ornithine, which is known to have better adhesion properties for neuronal cells as well as with brain derived nerve growth factors (BNDF) and nerve growth factor (NGF) which can modulate the growth and differentiation of neurons.Acknowledgements
This work was supported by a Research Grant (no. 27 (0243)/10/EMR-II, from Council of Scientific and Industrial Research (CSIR), India and Indian Institute of Science Education and Research-Kolkata (IISER-K), India as a start up Fund to JDS and RS. RS thanks Department of Science and Technology, New Delhi for Ramanujan Fellowship. We thank IISER-K and Department of Biotechnology, India for providing the fellowship and research support to AK. We thank CSIR for providing the fellowship and research support to NVK. We also thank University Grant Commission (UGC) for providing the research support to KB. Authors thank Mr Subhajit Das Sarma for his assistance in graphical work. Authors also thank IISER-K Microscopy facility and Mr Ritabrata Ghosh for his assistance in Microscopy.Notes and references
- E. Jan and N. Kotov, Nano Lett., 2007, 7, 1123–1128 CrossRef CAS PubMed.
- J. Das Sarma, Interdiscip. Perspect. Infect. Dis., 2010, 109239–109267 Search PubMed.
- J. Das Sarma, K. Iacono, L. Gard, R. Marek, L. C. Kenyon, M. Koval and S. R. Weiss, J. Virol., 2008, 82, 5519–5526 CrossRef CAS PubMed.
- J. Das Sarma, E. Scheen, S. H. Seo, M. Koval and S. R. Weiss, J. NeuroVirol., 2002, 8, 381–391 CrossRef CAS PubMed.
- J. Das Sarma, L. C. Kenyon, S. T. Hingley and K. S. Shindler, J. Neurosci., 2009, 29, 10272–10280 CrossRef CAS PubMed.
- J. H. Noseworthy, C. Lucchinetti, M. Rodriguez and B. G. Weinshenker, N. Engl. J. Med., 2000, 343, 938–952 CrossRef CAS PubMed.
- M. Sospedra and R. Martin, Annu. Rev. Immunol., 2005, 23, 683–747 CrossRef CAS PubMed.
- L. Steinman, R. Martin, C. Bernard, P. Conlon and J. R. Oksenberg, Annu. Rev. Neurosci., 2002, 25, 491–505 CrossRef CAS PubMed.
- B. D. Trapp, J. Peterson, R. M. Ransohoff, R. Rudick, S. Mork and L. Bo, N. Engl. J. Med., 1998, 338, 278–285 CrossRef CAS PubMed.
- E. Lavi, P. S. Fishman, M. K. Highkin and S. R. Weiss, Lab. Invest., 1988, 58, 31–36 CAS.
- I. Tsunoda, Y. Iwasaki, H. Terunuma, K. Sako and Y. Ohara, Acta Neuropathol., 1996, 91, 595–602 CrossRef CAS.
- J. J. Phillips, M. M. Chua, G. F. Rall and S. R. Weiss, Virology, 2002, 301, 109–120 CrossRef CAS.
- R. G. Tremblay, M. Sikorska, J. K. Sandhu, P. Lanthier, M. Ribecco-Lutkiewicz and M. Bani-Yaghoub, J. Neurosci. Methods, 2010, 186, 60–67 CrossRef CAS PubMed.
- E. B. Malarkey and V. Parpura, Acta Neurochirurgica Supplement, 2010, 106, 337–341 Search PubMed.
- M. P. Mattson, R. C. Haddon and A. M. Rao, J. Mol. Neurosci., 2000, 14, 175–182 CrossRef CAS.
- Y. Ni, H. Hu, E. B. Malarkey, B. Zhao, V. Montana, R. C. Haddon and V. Parpura, J. Nanosci. Nanotechnol., 2005, 5, 1707–1712 CrossRef CAS PubMed.
- H. Hu, Y. Ni, S. K. Mandal, V. Montana, B. Zhao, R. C. Haddon and V. Parpura, J. Phys. Chem. B, 2005, 109, 4285–4289 CrossRef CAS PubMed.
- N. W. Kam, E. Jan and N. A. Kotov, Nano Lett., 2009, 9, 273–278 CrossRef CAS PubMed.
- V. N. Rao, A. Kishore, S. Sarkar, J. Das Sarma and R. Shunmugam, Biomacromolecules, 2012, 13, 2933–2944 CrossRef PubMed.
- K. S. Kosik, L. D. Orecchio, L. Binder, J. Q. Trojanowski, V. M. Lee and G. Lee, Neuron, 1988, 1, 817–825 CrossRef CAS.
- L. I. Binder, A. Frankfurter and L. I. Rebhun, Ann. N. Y. Acad. Sci., 1986, 466, 145–166 CrossRef CAS PubMed.
- E. Debus, K. Weber and M. Osborn, Differentiation, 1983, 25, 193–203 CrossRef CAS PubMed.
- K. S. Shindler, D. Chatterjee, K. Biswas, A. Goyal, M. Dutt, M. Nassrallah, R. S. Khan and J. Das Sarma, J. Neuropathol. Exp. Neurol., 2011, 70, 470–480 CrossRef PubMed.
- D. M. Lawrence, C. E. Patterson, T. L. Gales, J. L. D'Orazio, M. M. Vaughn and G. F. Rall, J. Virol., 2000, 74, 1908–1918 CrossRef CAS PubMed.
- D. Edgar, R. Timpl and H. Thoenen, EMBO J., 1984, 3, 1463–1468 CAS.
- B. Grimpe, S. Dong, C. Doller, K. Temple, A. T. Malouf and J. Silver, J. Neurosci., 2002, 22, 3144–3160 CAS.
- H. K. Kleinman, G. C. Sephel, K. Tashiro, B. S. Weeks, B. A. Burrous, S. H. Adler, Y. Yamada and G. R. Martin, Ann. N. Y. Acad. Sci., 1990, 580, 302–310 CrossRef CAS PubMed.
- M. Nomizu, Y. Kuratomi, M. L. Ponce, S. Y. Song, K. Miyoshi, A. Otaka, S. K. Powell, M. P. Hoffman, H. K. Kleinman and Y. Yamada, Arch. Biochem. Biophys., 2000, 378, 311–320 CrossRef CAS PubMed.
- S. K. Powell, J. Rao, E. Roque, M. Nomizu, Y. Kuratomi, Y. Yamada and H. K. Kleinman, J. Neurosci. Res., 2000, 61, 302–312 CrossRef CAS.
- E. M. Harnett, J. Alderman and T. Wood, Colloids Surf., B, 2007, 55, 90–97 CrossRef CAS PubMed.
- J. A. Kim, N. Lee, B. H. Kim, W. J. Rhee, S. Yoon, T. Hyeon and T. H. Park, Biomaterials, 2011, 32, 2871–2877 CrossRef CAS PubMed.
- P. M. George, A. W. Lyckman, D. A. LaVan, A. Hegde, Y. Leung, R. Avasari, C. Testa, P. M. Alexander, R. Langer and M. Sur, Biomaterials, 2005, 26, 3511–3519 CrossRef CAS PubMed.
- L. W. Enquist, M. J. Tomishima, S. Gross and G. A. Smith, Vet. Microbiol., 2002, 86, 5–16 CrossRef CAS.
- S. E. Antinone and G. A. Smith, J. Virol., 2010, 84, 1504–1512 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |