
Synthesis, characterization and properties of a bio-based elastomer: polymyrcene†
Preetom Sarkara and
Anil K. Bhowmick*b
aDepartment of Chemistry, School of Basic Science, Indian Institute of Technology Patna, Patna - 800013, India
bRubber Technology Centre, Indian Institute of Technology Kharagpur, Kharagpur - 721302, India. E-mail: anilkb@rtc.iitkgp.ernet.in; anilbhowmick@gmail.com; Fax: +91 3222 220312; Tel: +91 3222 283180
First published on 7th November 2014
Abstract
An environmentally benign emulsion polymerisation technique was exploited to prepare a bio based polymer from a naturally occurring monoterpene, β-myrcene. The structure of the polymer was ascertained by spectroscopic measurements. Density functional theory was employed to determine the ground state optimised structure of β-myrcene. The persulfate initiated polymyrcene possesses 3,4 and 1,2 vinyl defects along with 1,4 microstructures, whereas the redox analogue contains solely 1,4 addition products. The synthesised polymer displayed a substantially high molecular weight (upto 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 860 Da for persulfate polymyrcene) and subzero (−73 °C) glass transition temperature along with shear thinning behaviour within the experimental conditions, thereby rendering itself as a promising entrant in the domain of bio-based elastomers.
860 Da for persulfate polymyrcene) and subzero (−73 °C) glass transition temperature along with shear thinning behaviour within the experimental conditions, thereby rendering itself as a promising entrant in the domain of bio-based elastomers.
Introduction
The outbreak of synthetic elastomer technology and elastomers as a strategic material is well documented in the history of World War II. The basic building blocks of synthetic elastomers are either derived from petrochemical resources (fossil fuel) or part of them contains petro-based synthons. As these resources dwindle with time, along with their price volatility and environmentally adverse effects, the scientific community is in dire search of a suitable replacement of the current petro based elastomers by sustainable alternatives.1 This paradigm shift of surge is reflected in the announcement of the Genencor and Amyris company to launch BioIsoprene® as a replacement for its petro based analogue.2,3 Thus, it is evident that the impetus for growth in this domain will surely rely on the development of environmentally benign methodologies which utilise abundant bio-resources.Nature produces rubber latex in over two thousand plant species by bio-synthetic (isopentenyl pyrophosphate) route.4 On the other hand, terpenes or terpenoids refer to one of the largest families of naturally occurring compounds and are secondary metabolites synthesized mainly by plants, particularly conifers.5,6 Though most terpenes share isoprene (2-methyl-1,4-butadiene) as an elementary unit (‘isoprene rule’),7 nature does not produce any polymer out of it. Thus, due to their abundance in nature, there has been great interest in producing polymers with terpenes as either functional entities or as the main constituent.8 To make use of the chemical functionalities (unsaturation, functional groups) present in terpene molecules, various chemical strategies and a wide range of polymerisation techniques9–19 had been studied. Unlike the terpenes used in the aforementioned reports, the acyclic monoterpene: 7-methyl-3-methylene-1,6-octadiene or β-myrcene resembles the classical chemistry of other petro based unsaturated hydrocarbons. β-myrcene is a component of essential oil of various plants like wild thyme, ylang–ylang, bay, cannabis, parsley, hops and its use as a fragrance and flavours are known from antiquity.20 Commercially, β-myrcene is prepared from the pyrolysis of β-pinene. Its use as a potential diesel fuel additive after hydrogenation is also reported in literature.21 Interestingly, very little thrust has been given to exploit this natural product in the field of elastomer and tyre technology. Kobayashi et al.22 prepared poly(3-methylenecyclopentene) using a combination of ring-closing metathesis and cationic polymerization starting from myrcene. Loughmari et al.23 reported co-ordination polymerisation of β-myrcene. Marval and Hwa24 studied the polymerisation of myrcene using Ziegler-type catalyst. Recently, Choi and Ritter25 reported a free radical copolymerisation of myrcene with diethyl fumarate and styrene, utilising water soluble β-cyclodextrin complex. All of the methodologies mentioned essentially rely on either utilisation of volatile organic compounds (especially solvents) or specialised techniques (catalyst), and were not carried out with application in mind. Thus, despite some studies on terpene-based polymers, the increasing leap for environmentally benign methodologies demands further development and implementation of a green polymerisation technique in this domain which will supplement the dearth of bio based synthetic elastomer. Being a solvent less and mature system along with its multitude of advantages (faster rate, high molecular weight products), emulsion polymerisation26,27 is cynosure in this area.
With our quest to develop a bio-based elastomer by employing a green technique, we herein report the persulfate as well as redox initiated emulsion polymerisation of β-myrcene and intend to give an insight into the chemical structure and the properties of the synthesised polymer. Vulcanisate properties of polymyrcene was first reported by Johanson et al.,28 although there was no work on the structure or characterisation or optimisation of the experimental conditions to obtain the best properties of the pristine polymer. Surprisingly, this polymer did not find attention for a long time after the preliminary investigation. We feel that further studies should be done to exploit its commercial viability in view of recent focus on sustainable and green elastomer. To the best of our knowledge, this is the first report of its kind which presents an in-depth analysis of the microstructure of the as obtained polymer using environmentally benign emulsion polymerisation technology along with theoretical calculation and elucidation of properties. The optimised structure of β-myrcene was determined using Density Functional Theory (DFT) calculation. The reactivity of the molecule was ascertained with the help of frontier molecular orbital theory and electrostatic potential (ESP) mapping. Thermal stability, various transitions and mechanical and dynamic mechanical properties of the synthesised polymer were measured and the findings were correlated with the microstructure. Furthermore, the particle nature of the emulsion latex was also reported.
Experimental
Materials
β-Myrcene (99%) was purchased from Sigma Aldrich company and the inhibitor, butylated hydroxytoluene was removed by shaking with 2(M) NaOH solution. Sodium dodecyl sulphate (SDS, 99%) was procured from Loba Chemie, India and used as received. Ammonium persulphate (APS, 98%) and sodium bicarbonate (NaHCO3, 99%) were obtained from E. Merck (India). Ferrous sulphate heptahydrate (FeSO4·7H2O, 98%) was purchased from Alfa Aesar India. Ethylenediaminetetraacetic acid sodium salt, potassium oleate, potassium phosphate tribasic (K3O4P), potassium chloride (KCl), sodium hydroxymethane sulfinate (SHS) and tert-butyl hydroperoxide solution (Luperox® TBH70X), were purchased from Sigma Aldrich company and used without further purification. All other chemicals were reagent-grade commercial products and used as received without further purification. Deionised water (DI) was taken for all the experiments.Persulfate initiated emulsion polymerisation of β-myrcene
Persulfate initiated emulsion polymerisation of β-myrcene was carried out following the recipe mentioned in Table 1. The general procedure followed for the emulsion polymerisation was as follows: at first, the emulsifier (SDS), DI water and buffer (NaHCO3) were added into the round bottom flask and mixed thoroughly at 250 rpm for 20 min to form the micelle. Thereafter, the β-myrcene monomer was added slowly over a period of 10 min and left as such for a further 20 min so as to obtain a stable emulsion. Then, the reactor was flushed twice with nitrogen and the temperature was set to 70 °C. Aqueous solution of the initiator (APS) was added into the reaction mixture drop-wise over 20 min in two intervals. The reaction was continued for 20 h to obtain a stable latex. Initially, the colour of the reaction mixture was off white, but with the progress of the reaction a light blue tinge appeared due to the particle growth in the emulsion medium (Tyndall effect). After the stipulated time, the latex was coagulated using excess acidified ethanol with continuous stirring. The obtained polymyrcene (PMy) was taken out, washed profusely with DI water to remove the traces of emulsifier and subsequently dried at 50 °C in vacuum for 24 h. The polymerisation is shown in Scheme 1. Polymerisation was also carried out at different temperatures (60–80 °C) and different times (4–24 h). | ||
| Scheme 1 Emulsion polymerisation of β-myrcene. | ||
Redox initiated emulsion polymerisation of β-myrcene
According to the recipe furnished in Table 2, redox initiated emulsion polymerisation was performed following strict addition sequence. At first, DI water, potassium oleate, potassium chloride and potassium phosphate tribasic buffer were charged into a round bottom flask and stirred at 250 rpm for 20 min. Subsequently, β-myrcene was introduced into the flask and mixed thoroughly for a further 20 min. Upon obtaining a stable emulsion, SHS, FeSO4 and Na-EDTA were added to the mixture. Thereafter, the reactor was flushed with nitrogen to remove traces of air, followed by the injection of TBHP solution into the flask. The polymerisation was allowed to proceed for 20 h at 25 °C. The obtained latex was then coagulated using excess acidified ethanol. The as obtained polymer was washed profusely with DI water and dried in vacuum oven at 50 °C for 24 h.| Ingredients | Amount (g, in phra) |
|---|---|
| a Parts per hundred parts of rubber. | |
| Monomer | 100 |
| DI water | 180 |
| Potassium oleate | 4.5 |
| Potassium chloride | 0.3 |
| Potassium phosphate tribasic | 2.0 |
| tert-Butyl hydroperoxide solution (TBHP) | 0.06 |
| Ferrous sulphate heptahydrate (FeSO4, 7H2O) | 0.01 |
| Ethylenediaminetetraacetic acid sodium salt | 0.05 |
| Sodium hydroxymethane sulfinate (SHS) | 0.05 |
Measurements and characterisation
Polymer molecular weight was determined by Agilent PL-GPC 50 instrument, using PLgel 5 μm Mixed-D column equipped with a refractive index detector and tetrahydrofuran as the eluent (sample concentration 1 mg ml−1). The GPC data were recorded at 25 °C at a flow rate of 1 ml min−1 (polystyrene standards were used for calibration). In order to determine the gel fraction of the synthesised polymers, the samples were extracted using THF under reflux condition for 8 h. The gel percentage was calculated as the ratio of dried polymer weight to its original value.The FT-IR spectra were recorded in Universal Attenuated Total Reflectance mode (UATR) using Perkin Elmer Spectrum 400 machine in a spectral range of 4000–650 cm−1 with a total of 8 scans per sample (resolution 4 cm−1).
1H NMR and 13C NMR spectrum were recorded in a AVANCE III 400 Ascend Bruker instrument operating at 400 MHz. CDCl3 was used as a solvent and the chemical shift values were reported in δ (ppm) relative to the 1H signals from the internal standard tetramethylsilane (TMS). The peak for deuterated solvent appeared at δ = 7.19 ppm for 1H NMR and δ = 77.22 ppm for 13C NMR.
Raman spectroscopy was recorded using an STR 750 series (Seki Technotron/Technos Instrument India) Raman spectrometer with a 633 nm He–Ne laser source and a grating of 600 lines per mm. The samples were placed on the sample stage and the laser focused on a specific area of the samples using 50× objective lens of the attached microscope (Olympus BX-51). The results reported are an average of data collected over 5 different points.
Density functional theory (DFT) calculation was carried out using Gaussian-09 software. The geometry (optimised structure) and frequency of β-myrcene were calculated with Becke's three-parameter hybrid functional (B3LYP) method by using 6-311G (d, p) as basis set. The electrostatic potential map (ESP) density of the optimised structure was generated using ArgusLab 4.0.1 software.
Differential scanning calorimetry measurements were recorded in a Perkin Elmer DSC8000 under nitrogen atmosphere. The following heating protocol was followed: the sample was heated from −96 °C to +100 °C, equilibrating at +100 °C for 2 min, cooling from +100 °C to −96 °C, equilibrating at −96 °C for 2 min, reheating from −96 °C to +100 °C. The heating or cooling rates were 5 °C min−1 in all the cases. To eliminate the thermal history, transition temperature was reported from the second heating run. Thermogravimetric analysis were carried out using SDT Q600 (TA Instruments) under nitrogen flow at a ramp rate of 10 °C min−1.
The dynamic mechanical properties of synthesised polymer were measured using a DMA-Q800 machine in tension mode. All the samples (11 × 6.3 × 0.24 mm) were analyzed at a constant frequency of 1 Hz, at a heating rate of 3° C min−1 and strain amplitude of 30 μm over a temperature range of −100 °C to +100 °C. Because of low strength of the synthesised polymer, the glass fibre cloth was impregnated with a solution of the polymer in chloroform and a thin film of uniform thickness was deposited on it followed by drying at room temperature. Glass fibre cloth was chosen as it did not show a glass transition within the temperature range of interest.29 Only the Tan Delta peak was taken into account from this experiment.
The rheological measurements were performed using a MCR302 Anton Paar Modular Compact Rheometer having parallel plate geometry of 8 mm diameter and with a 1.1 mm gap in between the plates. Frequency sweep experiments were conducted at 30 °C and at a constant strain of 0.2% in the frequency range between 0.1 to 10 Hz. Temperature sweep experiments were carried out at 0.2% constant strain and 1 Hz frequency in between −10 °C to +100 °C temperature employing a ramp rate of 3 °C/min.
The stress–strain behaviour of the synthesised polymyrcene was recorded using Tinius Olsen H5KS UTM machine with a 5 N load cell at room temperature and a cross-head speed of 25 mm min−1. The samples for tensile testing were prepared by solution casting technique. As the samples were weak, attempt to prepare the dog-bone type tensile specimen by using standard ASTM (D412) cutter created wrinkles and defects in the samples. Hence, it was decided to prepare rectangular samples.30 The dimensions of the rectangular tensile specimen were: length = 35 mm, width = 7.5 mm, thickness = 0.5 mm.
Wide angle X-ray diffraction analysis was carried out using Rigaku TT RAX3 XRD machine in the range of 2θ from 10°–50° (scan rate 3° per min) with CuKα (0.154 nm) as radiation source at 50 kV and 100 mA.
The morphology of the latex was characterised using field emission scanning electron microscope (Hitachi, S-4800 FESEM) at an accelerating voltage of 10 kV. The latex was diluted with water and sonicated prior to casting and drying on silicon wafer and thereafter coated with gold by using Hitachi E-1010 ion sputter system. The particle nature of the latex was analysed by dynamic light scattering (DLS) method using Delsa® Nano Submicron Particle Size and Zeta Potential Particle Analyzer PN A54412 from Beckman Coulter with a laser as a light source and a detector that detected the scattered light at an angle of 165°.
All the experimental results on characterisation and polymerisation were based on three measurements.
Results and discussion
Synthesis of polymyrcene (PMy) by emulsion polymerisation
Ammonium persulfate initiated emulsion polymerisation of β-myrcene was performed at 70 °C at atmospheric pressure. High temperature was maintained to furnish sufficient free radicals for the polymerisation process. Sodium dodecyl sulphate (SDS) was used as a surfactant and sodium bicarbonate was used as a buffer. Based on the polymer molecular weight and percentage yield, the optimised reaction conditions (polymerisation time and reaction temperature) for persulfate initiated process were frozen, and all the analysis was carried out using the polymer synthesised at the optimised set of reaction conditions. To facilitate the polymerisation process at room temperature, redox emulsion technique was also carried out. Na-EDTA was used as a sequestering agent, sodium hydroxymethane sulfinate (SHS) was used as an oxidant, ferrous sulphate heptahydrate (FeSO4, 7H2O) as a reductant and tert-butyl hydroperoxide (TBHP) as a free radical generator.Time and temperature dependence of PMy synthesis
In order to investigate the effect of time and temperature on the PMy synthesis, the persulfate initiated emulsion polymerisation was carried out at first at five different temperatures of 5 °C interval and for a fixed reaction time of 20 h. The lower and upper limit of the temperature is such chosen to ensure complete initiator decomposition in the former case (well above the half life temperature of the initiator species) and preventing destabilisation of the dispersion medium (i.e. water) of the polymerisation system respectively. Fig. 1a shows that with increasing temperature, both molecular weight and percentage yield exhibit increasing trend upto 70 °C, after which a reversion trend is followed. This can be attributed to the fact that with increase in temperature, the propagation rate of the reaction increases; but at high temperature, thermal decomposition of the polymer chains predominates, thereby reducing the molecular weight and percentage yield value. Thus, 70 °C was chosen as the optimum reaction temperature. To study the effect of time on the polymerisation reaction, aliquots were taken out from the reaction mixture at 4 h time interval and upto 24 hours followed by GPC measurements and yield calculation. | ||
| Fig. 1 (a) Temperature dependence of PMy synthesis on yield and molecular weight, (b) time dependence of PMy synthesis on yield and molecular weight. | ||
It is evident from Fig. 1b that percentage yield increases with reaction time and the molecular weight reaches its maximum value of 92860 Da at 20 h. In line with the temperature effect, at higher reaction time, chain scission occurs which produces oligomers and hence there is a decreasing trend of molecular weight at long reaction time.27 Therefore, from the cumulative study of temperature and time dependence, the optimum reaction condition for the persulfate initiated emulsion polymerisation was fixed at 70 °C for 20 h. Table 3 summarises the molecular weight and yield values for the synthesised PMy at different conditions. Redox initiated polymerisation yields polymer with high molecular weight in comparison to persulfate initiated one (Table 3). This is because the redox reaction was carried out at room temperature, and the side reactions and chain transfer effects are suppressed which facilitate the production of high molecular weight polymer. Due to micro crosslinking between the growing chains (two nos of residual unsaturation in the polymer structure), the gel content of the synthesised polymer increases with increasing reaction temperature and time. For the redox system, the high gel content value is in good agreement with other studies.25
| Sample | Time (h) | Temperature (°C) | Mn (Da) | PDI | % Gel | % Yield |
|---|---|---|---|---|---|---|
| a Values in the parenthesis indicate the standard deviation based on three measurements. | ||||||
| PMy60 °C | 20 | 60 | 43270 (±250)a |
2.64 | 5 | 83.0 (±0.4) |
| PMy65 °C | 20 | 65 | 73570 (±280) |
2.51 | 7 | 87.0 (±0.3) |
| PMy70 °C | 20 | 70 | 92860 (±300) |
2.48 | 12 | 96.0 (±0.6) |
| PMy75 °C | 20 | 75 | 88650 (±590) |
2.42 | 14 | 91.0 (±0.8) |
| PMy80 °C | 20 | 80 | 85850 (±490) |
2.55 | 14 | 83.0 (±0.4) |
| PMy4 h | 4 | 70 | 33970 (±490) |
1.77 | 2 | 87.0 (±0.3) |
| PMy8 h | 8 | 70 | 60580 (±220) |
2.25 | 5 | 89.0 (±0.3) |
| PMy12 h | 12 | 70 | 69450 (±500) |
2.27 | 8 | 91.0 (±0.6) |
| PMy16 h | 16 | 70 | 75140 (±380) |
2.26 | 9 | 93.0 (±0.5) |
| PMy20 h | 20 | 70 | 92860 (±300) |
2.48 | 12 | 96.0 (±0.4) |
| PMy24 h | 24 | 70 | 63550 (±450) |
3.02 | 12 | 98.0 (±0.5) |
| PMyredox | 20 | 25 | 109780 (±410) |
3.13 | 17 | 66.0 (±0.7) |
FT-IR and NMR characterisation of the synthesised PMy
Fig. 2 represents the FT-IR spectra of β-myrcene and PMy20 h. The weak peak at 3093 cm−1 in the monomer represents the![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretching of C1, C3 and C4, which disappears upon polymerisation. The broad absorption peaks at 2970, 2931 and 2860 cm−1 in the monomer are assigned to the –CH3, –CH2 and –CH asymmetric stretching vibration respectively, which get broadened due to the formation of more number of such groups in the polymer. The strong absorption peak at 1596 cm−1 is assigned to C1C2 and C3C4 stretching frequency (Fig. 2). Due to conjugation, this stretching appears at lower wave number value. A small peak at 1641 cm−1 can be ascribed to the presence of isolated C7C8 in the structure. The disappearance of the former CC peaks and simultaneous broadening of the latter one indicates the consumption of C1C2 and C3C4 during the polymerisation process followed by formation of C2C3 and simultaneous retention of the isolated unsaturation. The confinement of C–H bending peak for tri substituted alkene (here C7) at 824 cm−1 even after polymerisation reiterates that the isolated double bond is indeed preserved during polymerisation (which is evident from theoretical calculation discussed later). The characteristics absorption peaks around 1444 and 1376 cm−1 are due to the –CH2 and –CH3 bending vibration of C5–C6 and C9–C10 centres respectively. After polymerisation, generation of more methylene unit is reflected in the relative increment of the corresponding peak area at 1444 cm−1. The strong absorption peaks around 992 and 889 cm−1 in the monomer is due to the sp2 C–H bending vibration of C1, C3 and C4 centres respectively. While the sharp decrease in the intensities of these bands in the polymer is due to the consumption of these two unsaturations (C1C2 and C3C4) during polymerisation, the small residual peak in these two regions is due to the formation of 3,4 structure (C1C2 is preserved) and 1,2 vinyl (C3C4 is preserved) structure in the polymer as ascertained from the NMR analysis discussed in the next section. The small peak at 735 cm−1 is due to the –CH2 rocking vibration which is preserved in the polymer with lower intensity due to restriction of rocking movement in the macromolecular chains.
C–H stretching of C1, C3 and C4, which disappears upon polymerisation. The broad absorption peaks at 2970, 2931 and 2860 cm−1 in the monomer are assigned to the –CH3, –CH2 and –CH asymmetric stretching vibration respectively, which get broadened due to the formation of more number of such groups in the polymer. The strong absorption peak at 1596 cm−1 is assigned to C1C2 and C3C4 stretching frequency (Fig. 2). Due to conjugation, this stretching appears at lower wave number value. A small peak at 1641 cm−1 can be ascribed to the presence of isolated C7C8 in the structure. The disappearance of the former CC peaks and simultaneous broadening of the latter one indicates the consumption of C1C2 and C3C4 during the polymerisation process followed by formation of C2C3 and simultaneous retention of the isolated unsaturation. The confinement of C–H bending peak for tri substituted alkene (here C7) at 824 cm−1 even after polymerisation reiterates that the isolated double bond is indeed preserved during polymerisation (which is evident from theoretical calculation discussed later). The characteristics absorption peaks around 1444 and 1376 cm−1 are due to the –CH2 and –CH3 bending vibration of C5–C6 and C9–C10 centres respectively. After polymerisation, generation of more methylene unit is reflected in the relative increment of the corresponding peak area at 1444 cm−1. The strong absorption peaks around 992 and 889 cm−1 in the monomer is due to the sp2 C–H bending vibration of C1, C3 and C4 centres respectively. While the sharp decrease in the intensities of these bands in the polymer is due to the consumption of these two unsaturations (C1C2 and C3C4) during polymerisation, the small residual peak in these two regions is due to the formation of 3,4 structure (C1C2 is preserved) and 1,2 vinyl (C3C4 is preserved) structure in the polymer as ascertained from the NMR analysis discussed in the next section. The small peak at 735 cm−1 is due to the –CH2 rocking vibration which is preserved in the polymer with lower intensity due to restriction of rocking movement in the macromolecular chains.
 | ||
| Fig. 2 FT-IR spectra of β-myrcene and PMy20 h. | ||
1H and 13C NMR characterisation of the synthesised PMy
The 1H and 13C NMR (inset) spectra of β-myrcene are presented in Fig. 3a. Fig. 3b and c represents 1H and 13C NMR spectra of persulfate initiated polymyrcene (PMy20 h) respectively. The spectral signals are well assigned to various magnetically different protons and carbons. The structure of the monomer is much like an isoprene unit, except the methyl group is replaced by an aliphatic chain having an isolated unsaturation (i.e. C7C8). Thus, the polymer may have four different types of microstructure viz. 1,4-cis, 1,4-trans, 1,2 vinyl and 3,4-polymyrcene. The spectral assignments are in good accord with the available literature.23,31 The sequencing of the carbon atoms are adopted from the literature.23,31
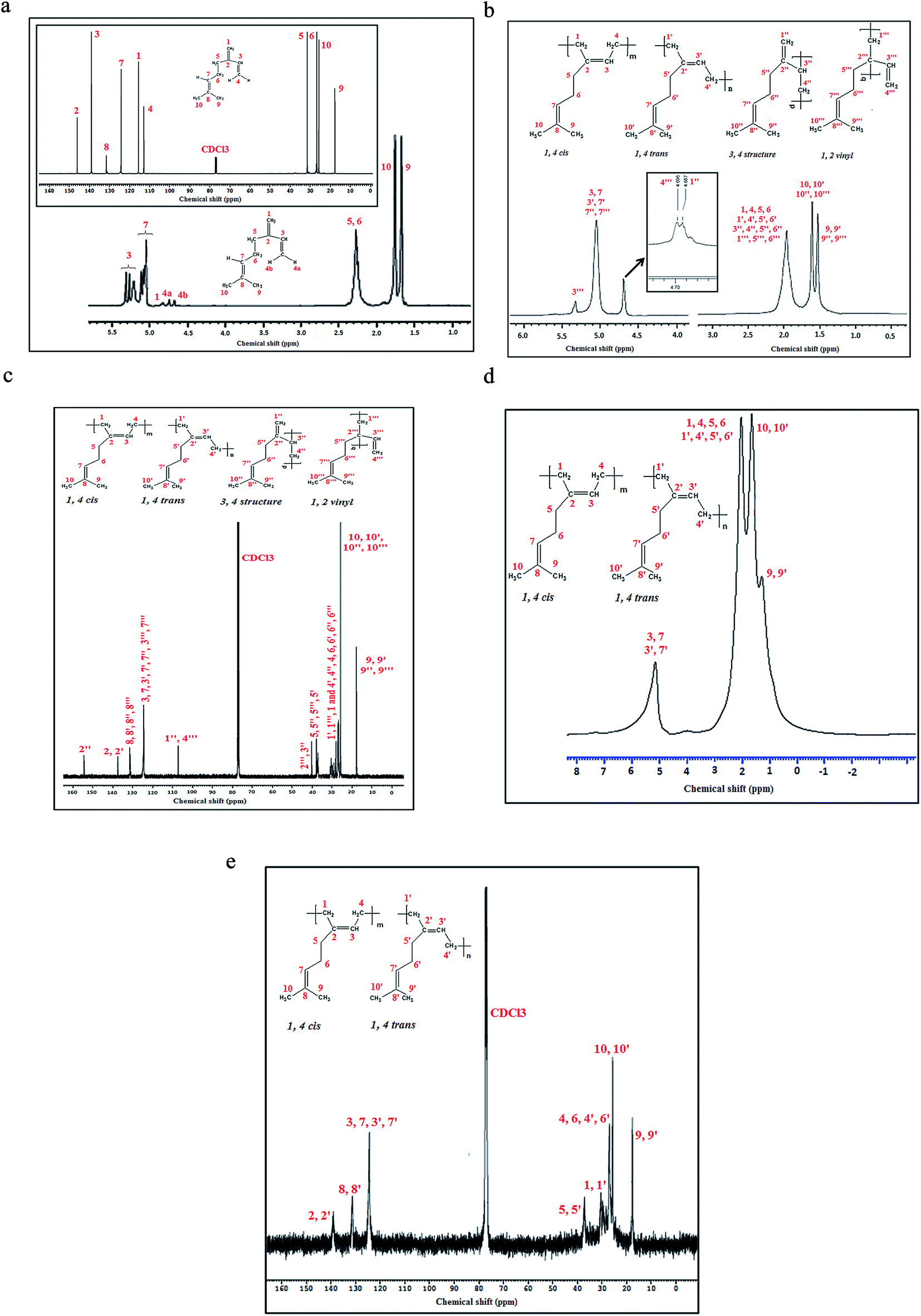 | ||
| Fig. 3 (a): 1H and 13C NMR spectra of β-myrcene, (b): 1H NMR spectrum of PMy20 h, (c): 13C NMR spectrum of PMy20 h, (d): 1H NMR spectrum of PMyredox, (e): 13C NMR spectrum of PMyredox. | ||
The protons of the two methyl groups attached to C8 in all the microstructures appear at upfield values (δ = 1.60 ppm and 1.52 ppm). The methylene protons of all the microstructures appear as a broad peak at 1.96 ppm. The upfield shift of all these protons relative to their monomer can be ascribed due to the generation of long chain macromolecules which shield each other from the external magnetic field (cf. Fig. 3a and b).
According to the literature report32,33 available on NMR studies of polyisoprene, the peaks above 4.5 ppm indicates the olefinic protons of 1,2 vinyl addition (5.32 ppm of 3′′′ H). A close look into the peak around 4.6 ppm reveals the presence of two splitting pattern at 4.69 ppm (4′′′ H of 1,2 vinyl) and 4.68 ppm (1′′ H of 3,4 structure). The olefinic CH– for both 1,4-cis, 1,4-trans as well as CH– of C7 appears as a single peak at 5.04 ppm. Taking these three peaks (δ = 5.04, 4.69 and 4.68 ppm) into account, it is estimated that the persulfate initiated polymyrcene consists of 47% 1,4-cis and 1,4-trans mixture, 29% 1,2 vinyl and 24% 3,4 structure. As no signature peak for 1,4-cis and 1,4-trans structure was obtained, exact percentage of these microstructures could not be determined.
The 13C NMR spectrum of persulfate initiated PMy shows several sharp peaks and few overlapped peaks in the region of δ = 25–30 ppm. Due to long macromolecular chains, the exact degeneracy of the peaks in 13C NMR spectra is very much unlikely. Peak at 154 ppm suggests the formation of 3,4 structure in the polymer. 2C and 2′C in -cis and -trans structures respectively appear at lower ppm (δ = 137.5 ppm), as they are part of the main chain of the polymer. The retention of peaks around 131.5 and 124.6 ppm in polymer (131.5 and 124.2 ppm respectively in monomer) indicates that the isolated unsaturation (C7C8) is indeed preserved and polymerisation took place at C1C2 and C3C4 position. The peak for 3C, 3′C, 3′′′C along with C7 appears at the same position (δ = 124.69 ppm). The vinyl carbons i.e. 1′′C (of 3,4 structure) and 4′′′C (of 1,2 vinyl) appeared as a single peak at 107.1 ppm. As in 1H NMR, the two methyl carbons attached to the C8 centre for all the microstructures were detected at up-field values (δ = 25.8 and 17.7 ppm). Due to the presence of attached hydrogen with 3′′ C, it showed an upfield value (δ = 39.9 ppm) compared to 2′′′C (δ = 40.37 ppm). Due to -cis structure, 5C is least shielded followed by 5′′C. The close proximity of the polymer chains with 5′C and 5′′′C leads to the higher shielding effect which reduces the chemical shift values. All other methylene protons (1, 4 and 6 Cs) appear in the range of 29.7–26.5 ppm and are overlapped with each other.
As shown in Fig. 3d, the 1H NMR spectrum of redox initiated polymyrcene shows a single peak at 5.14 ppm corresponding to olefinic protons (3,7,3′,7′ H) of CH– units. The absence of any other peak in the range of 4.5 to 5.5 ppm indicates that neither 3,4 structure nor 1,2 vinyl structure has formed and the microstructure predominantly contains 1,4 addition product (mixture of 1,4-cis and 1,4-trans). No distinct peak for all other methylene and methyl protons is observed, rather a bimodal peak (δ = 2.02 ppm and 1.64 ppm) followed by a hump (δ = 1.27 ppm) is obtained in the 1H NMR spectrum.
The 13C spectrum (Fig. 3e) of redox initiated polymyrcene also does not contain the signature peak of 2′′ C (of 3,4 structure) and 1′′,4′′′ C (of 3,4 structure and 1,2 vinyl structure respectively), indicating that the microstructure is devoid of 3,4 and 1,2 vinyl defects. Due to lower reaction temperature, the side reactions are suppressed in the case of redox polymerisation, thereby leading to polymyrcene that is rich in 1,4 microstructure. The chemical shift values of all the other methylene and methyl carbons are well in accordance with those of persulfate initiated polymyrcene.
Raman spectra of synthesised PMy
Raman spectra of β-myrcene and its polymer were recorded to compliment the observation of FTIR measurement. These are represented in Fig. 4. Raman band at 3006 cm−1 is assigned to the aliphaticC–H stretching mode of C1, C3 and C4 centre which gets diminished after polymerisation. The combined –CH2 and –CH3 asymmetric stretching bands appear as a broad band at 2913 cm−1 in the spectrum of the monomer which are intensified after polymerisation. Raman bands at 1632 and 1670 cm−1 in the monomer are ascribed to the presence of conjugated (C1C2 and C3C4) and isolated (C7C8) unsaturation respectively. It is anticipated that after polymerisation, the newly formed unsaturation (C2C3) and the isolated one (C7C8 as present in the monomer) appear as a single peak around 1668 cm−1. A broad band around 1435 cm−1 in the monomer is assigned for –CH2 bending which shifts to higher wave number value of 1444 cm−1 after polymerisation due to the formation of more such units, whereas the peak position due to –CH3 bending at 1381 cm−1 remains unaltered even after polymerisation (though its intensity gets enhanced probably due to macromolecular entanglement after polymerisation). The peaks at 1287 and 1327 cm−1 in the polymer are due to the wagging vibration of the methylene unit.
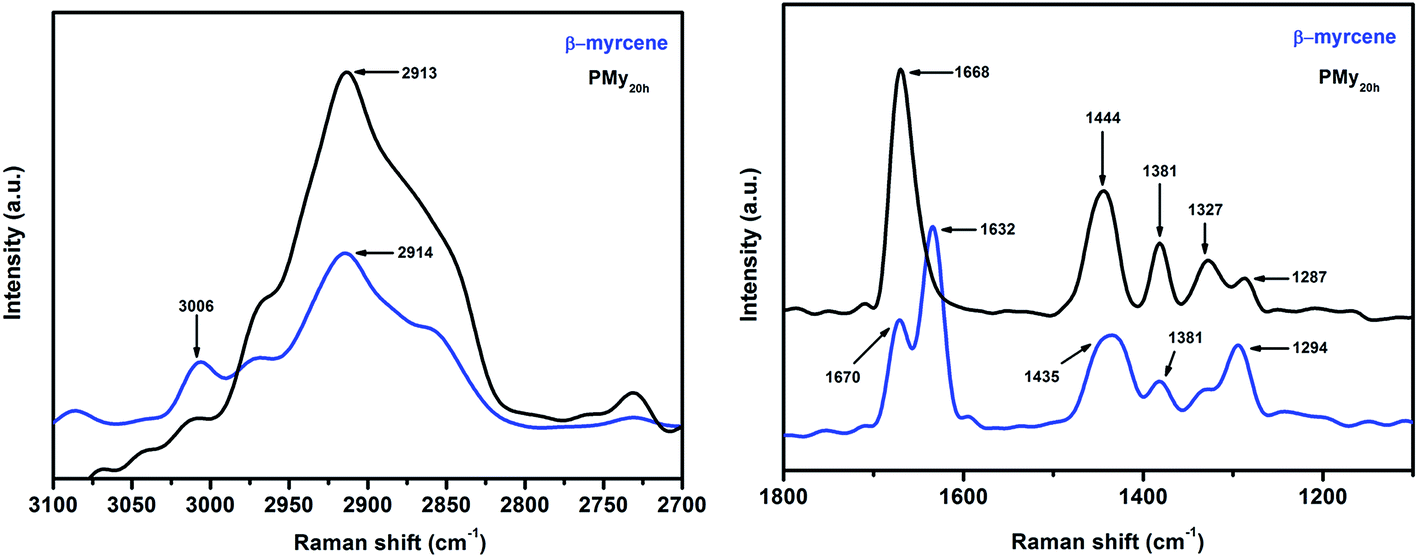 | ||
| Fig. 4 Raman spectra of β-myrcene and PMy20 h. | ||
Theoretical studies
In order to understand the relation between chemical structure and reactivity, and to provide an insight into the electronic properties, Density Functional Theory (DFT)34 calculation was performed with Becke's three-parameter hybrid functional (B3LYP) method using 6-311G (d, p) as a basis set. Calculations of molecular geometries and energy levels of polymers employing DFT is difficult due to their inherent macromolecular nature, but the calculation can be approximated using repeat unit of the same.35 Fig. 5a represents the optimised structure of β-myrcene molecule in the ground state computed by B3LYP/6-311G (d, p) level. The frontier orbitals of the same are also presented in Fig. 5b. It can be seen that though the HOMO is spread over the entire molecule, the LUMO is mainly localised onto the conjugated double bonds, i.e. C1C2 and C3C4 positions. Based on the B3LYP/6-311G (d, p) calculation, the energy of HOMO and LUMO is found to be −6.355 and −1.078 eV respectively, thereby indicating possible charge transfer from HOMO to LUMO and subsequent accumulation of charges on the conjugated double bond makes it very much avid towards polymerisation. The value of ground state dipole moment of β-myrcene calculated by B3LYP/6-311G (d, p) level is 0.4922 Debye. To envisage the charge distribution three dimensionally, electrostatic potential mapping (ESP) of the β-myrcene monomer was done. It can be used to evaluate the electronic distribution around the molecular surface and has been used35 for predicting sites and relative reactivity towards electrophilic attack. From the ESP mapping, it is clear that C1, C2, C3, C4 are surrounded by a greater density of negative charge, thus making this site potentially more prone towards polymerisation. Thus, combining the spectroscopic analysis and theoretical studies, it is evident that the polymerisation of β-myrcene proceeds via C1C2 and C3C4 pathways and the isolated unsaturation (C7C8) remains unaltered.
 | ||
| Fig. 5 (a) The ground state optimized structure of β-myrcene in B3LYP/6-311G (d, p), (b) the molecular orbitals and their energies of β-myrcene molecule in the ground state in B3LYP/6-311G (d, p), (c) electrostatic potential map for optimised β-myrcene molecule, high electrostatic potential: relative absence of electron; low electrostatic potential: relative abundance of electron. | ||
Wide angle X-ray diffraction analysis of the synthesised PMy
X-ray diffraction analysis was performed to study the crystalline nature of the synthesised polymyrcene. The XRD plot (Fig. S1†) of the synthesised polymer shows a broad hump at 2θ = 19°. Thus, the polymer film displayed a typical pattern of an amorphous polymer, much like a natural rubber. Branching at 2,2′,3′′,2′′′ C position in all of the microstructures prevents close packing of the chains, thereby rendering the polymer amorphous.Thermal analysis of the synthesised polymer
Glass transition temperature (Tg) of a polymer plays an important role in dictating whether a particular polymer is in the rubbery state at room temperature. Elastomers are amorphous in nature and thus should have a Tg much below the ambient temperature, preferably at sub-zero range. Fig. 6a shows the DSC thermograms of persulfate and redox initiated polymer. For persulfate initiated polymyrcene, the DSC thermogram of second heating run shows a distinct baseline shift at around −73 °C which corresponds to the glass transition temperature of the polymer. The transition temperature value is close to that of the natural rubber reported in literature,36 thereby implying rubber like behaviour of the synthesised polymer. The redox variety of the polymer showed inflexion point at −60 °C, approximately 13 °C higher than the persulfate initiated species. This shift of temperature to higher value can be attributed to both increase in molecular weight which increases entanglement, thereby restricting segmental mobility, higher gel content and 1,4 addition rich microstructure which facilitates dense packing compared to its persulfate initiated analogue. Fig. 6b represents the thermal decomposition curve of both persulfate and redox initiated polymyrcene. Unlike natural rubber, which shows a single degradation peak, the synthesised polymer exhibits two step degradation pattern. In the case of persulfate initiated polymyrcene, the sample weight gradually falls after onset of degradation, until it reaches ∼425 °C beyond which the polymer degrades off quickly. In the case of redox initiated polymer, it shows a plateau after first weight loss and falls off rapidly upon reaching ∼427 °C. As analysed from the NMR spectra, the persulfate initiated polymyrcene contains 3,4 and 1,2 vinyl defects in its microstructure in comparison to redox initiated polymyrcene which contains only 1,4 microstructure. The effect of microstructure on the degradation of polybutadiene rubber is documented in the literature.37 | ||
| Fig. 6 (a) DSC thermograms of persulfate and redox initiated polymyrcene, (b) TGA thermograms of persulfate and redox initiated polymyrcene. | ||
Rheological studies and mechanical properties of the synthesised polymer
The tanδ versus temperature plot for both persulfate and redox polymyrcene are presented in Fig. 7a. The tanδ peak and hence the glass transition temperature of the persulfate and redox polymyrcene was found to be around −53.3 and −50.0 °C respectively. The numerical values follow the same trend as in DSC analysis. The higher Tg's are due to dynamic heating rate in the experiment. The monotonous decrease of complex viscosity with increasing angular frequency for both the samples is evident from Fig. 7b. It is anticipated that due to high molecular weight and defect free microstructure (1,4 microstructure aids in much dense packing) the complex viscosity for redox polymyrcene shows such higher value.
 | ||
| Fig. 7 (a) tanδ overlay plot for persulfate and redox polymyrcene, (b) plot of complex viscosity versus angular frequency for persulfate and redox polymyrcene, (c) tensile stress–strain plot of synthesised polymyrcene (inset shows a representative strip of polymyrcene used in tensile measurements). | ||
In order to understand the flow behaviour of the synthesised polymer, Power law model38 was employed. Power law relates complex viscosity (η*) with angular frequency (ω) by flow consistency index (k) and flow behaviour index (n) by the following equation:
| η* = kω(n−1). | (1) |
After linear curve fitting, from the slope, flow behaviour index for both the polymers was found to be less than one (n < 1), indicating pseudoplastic nature (shear thinning behaviour).38 PMyredox, having higher ‘n’, is less shear thinning than PMy20 h. The tanδ value at 0 °C (indirect measure of wet skid resistance) and 60 °C (indirect measure of rolling resistance) for the synthesised polymer are presented in Table 4.
The tensile stress–strain plot of the synthesised polymer is presented in Fig. 7c. The strips were prepared by casting a solution of synthesised polymer in chloroform on Teflon® petri-dish and driving off the solvent at room temperature. The stress–strain curve for persulfate initiated polymyrcene (unvulcanised and gum) shows higher tensile strength value (97.8 kPa) and higher elongation (upto 60%) than the redox polymyrcene. It is anticipated that presence of 3,4 and 1,2 vinyl microstructure leads to higher physical entanglement in the case of persulfate polymyrcene which aids in higher elongation and tensile strength. The relatively high 1,4-trans content in redox polymyrcene hinders such phenomenon. Table 5 presents the mechanical properties of the polymyrcene polymers. These values are similar to other unvulcanised and gum rubbers.39 Hence, this elastomer may find application as a single rubber, in polymer blends, composites as well as in adhesives.
Analysis of particle morphology of the latex emulsion
To analyse the particle nature of the latex, dynamic light scattering method was used and the particle nature of the latex was further confirmed from FESEM images. The particle size distribution and mean particle size of the polymyrcene latex taken at different time intervals are presented in Fig. S2.† The particle size increases from 52.6 nm at 4 h to 72.1 nm at 24 h at 70 °C. The FESEM images (Fig. S3†) were taken by drop casting the latex emulsion on silicon wafer and subsequently driving off the water. Due to the removal of the dispersion medium, the particles tend to coalesce with each other, thereby becoming prone to form larger aggregates (black circles in Fig. S3†). However, we measured the diameter of the nodules in the aggregates. The average diameter is in accord with the value obtained from DLS measurements.Conclusions
In the present work, we have successfully synthesised a bio-based elastomer from naturally occurring monoterpene, β-myrcene, utilising emulsion polymerisation technique. The polymyrcene synthesised following the optimised set of reaction conditions (at 70 °C for 20 h) displayed reasonably high molecular weight of 92860 Da and a subzero glass transition temperature of −73 °C. From the 1H and 13C NMR analysis, it was found that the persulfate initiated polymyrcene contained 1,2 vinyl and 3,4 defects along with 1,4 cis and 1,4 trans microstructure. The redox analogue showed number average molecular weight of 1,09780 Da. The absence of peak in the range of δ = 4.5 to 5.5 ppm in the 1H NMR spectrum indicated that it contained predominately 1,4 microstructures. Spectroscopic measurements coupled with DFT calculation confirmed the participation of conjugated double bond in the polymerisation process and simultaneous conservation of the isolated one. The complex viscosity data was fitted to Power Law model and the finding revealed the pseudoplastic behaviour of the synthesised polymer. The amorphous nature, glass transition temperature and thermal stability of the synthesised polymyrcene were found to be similar to those of other rubbers, thus paving the way for its application as an elastomer of the future.
Acknowledgements
Part of the work and analysis of the results were carried out at Indian Institute of Technology Kharagpur. PS would like to acknowledge the facilities provided by IIT Kharagpur.References
- A. Gandini, Polymers from Renewable Resources: A Challenge for the Future of Macromolecular Materials, Macromolecules, 2008, 41, 9491 CrossRef CAS
.
- R. Singh, Facts, Growth and Opportunities in Industrial Biotechnology, Org. Process Res. Dev., 2010, 15, 175 CrossRef
- J. Yang, G. Zhao, Y. Sun, Y. Zheng, X. Jiang, W. Liu and M. Xian, Bio-isoprene Production Using Exogenous MVA Pathway and Isoprene Synthase in Escherichia coli, Bioresour. Technol., 2012, 104, 642 CrossRef CAS PubMed
- A. K. Bhowmick and H. L. Stephens, Handbook of Elastomers, Marcel Dekker AG, New York, 2001, ch. 2 Search PubMed
- M. N. Belgacem and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, Elsevier, Amsterdam, 2008, ch. 2 Search PubMed
- W. F. Erman, Chemistry of Monoterpenes, Marcel Dekker, New York, 1985 Search PubMed
- I. L. Finar, Organic Chemistry Vol. 2, Stereochemistry and The Chemistry of Natural Products, Longmans Green and Co. Ltd., London, 1964, ch. 8 Search PubMed
- P. A. Wilbon, F. Chu and C. Tang, Progress in Renewable Polymers from Natural Terpenes, Terpenoids and Rosin, Macromol. Rapid Commun., 2013, 34, 8 CrossRef CAS PubMed
- M. Firdaus, M. de Espinosa Lucas and A. R. Meier Michael, Terpene-Based Renewable Monomers and Polymers via Thiol-Ene Additions, Macromolecules, 2011, 44, 7253 CrossRef CAS
- A. Corma, S. Iborra and A. Velty, Chemical Routes for the Transformation of Biomass into Chemicals, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed
- E. Grau and S. Mecking, Polyterpenes by Ring Opening Metathesis Polymerization of Caryophyllene and Humulene, RSC Green Chem. Ser., 2013, 15, 1112 RSC
- S. Sharma and A. K. Srivastava, Synthesis and Characterization of Copolymers of Limonene with Styrene Initiated by Azobisisobutyronitrile, Eur. Polym. J., 2004, 40, 2235 CrossRef CAS PubMed
- M. Firdaus and A. R. Meier Michael, Renewable Polyamides and Polyurethanes Derived from Limonene, RSC Green Chem. Ser., 2013, 15, 370 RSC
- J. E. Puskas, A. L. Gergely and G. Kaszas, Controlled/Living Carbocationic Copolymerization of Isobutylene with Alloocimene, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 29 CrossRef CAS
- Y. Shi, G. Shan and Y. Shang, Role of Poly(ethylene glycol) in Surfactant-free Emulsion Polymerization of Styrene and Methyl methacrylate, Langmuir, 2013, 29, 3024 CrossRef CAS PubMed
- K. Satoh, H. Sugiyama and M. Kamigaito, Biomass Derived Heat Resistant Alicyclic Hydrocarbon Polymers: Poly(terpenes) and Their Hydrogenated Derivatives, RSC Green Chem. Ser., 2006, 8, 878 RSC
- N. A. Kukhta, I. V. Vasilenko and S. V. Kostjuk, Room Temperature Cationic Polymerization of β-pinene Using Modified AlCl3 Catalyst: Toward Sustainable Plastics From Renewable Biomass Resources, RSC Green Chem. Ser., 2011, 13, 2362 RSC
- M. Matsuda, K. Satoh and M. Kamigaito, 1:2-Sequence-Regulated Radical Copolymerization of Naturally Occurring Terpenes with Maleimide Derivatives in Fluorinated Alcohol, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1774 CrossRef CAS
- M. H. Alvès, H. Sfeir, F. Tranchant Jean, E. Gombart, G. Sagorin, S. Caillol, L. Billon and M. Save, Terpene and Dextran Renewable Resources for the Synthesis of Amphiphilic Biopolymers, Biomacromolecules, 2014, 15, 242 CrossRef PubMed
- A. Behr and L. Johnen, Myrcene as a Natural Base Chemical in Sustainable Chemistry: A Critical Review, ChemSusChem, 2009, 2, 1072 CrossRef CAS PubMed
- N. I. Tracy, D. Chen, D. W. Crunkleton and G. L. Price, Hydrogenated Monoterpenes as Diesel Fuel Additives, Fuel, 2009, 88, 2238 CrossRef CAS PubMed
- S. Kobayashi, C. Lu, R. Hoye Thomas and M. A. Hillmyer, Controlled Polymerization of a Cyclic Diene Prepared from the Ring-Closing Metathesis of a Naturally Occurring Monoterpene, J. Am. Chem. Soc., 2009, 131, 7960 CrossRef CAS PubMed
- S. Loughmari, A. Hafid, A. Bouazza, E. Bouadili Abdelaziz, P. Zinck and M. Visseaux, Highly Stereoselective Coordination Polymerization of β-Myrcene from a Lanthanide-Based Catalyst: Access to Bio-Sourced Elastomers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2898 CrossRef CAS
- C. S. Marval and C. C. L. Hwa, Polymyrcene, J. Polym. Sci., 1960, XLV, 25 CrossRef
- S. W. Choi and H. Ritter, Novel Polymerization of Myrcene in Aqueous Media via Cyclodextrin-Complexes, e-Polymers, 2007, 45, 1 Search PubMed
- A. van Herk, Chemistry and Technology of Emulsion Polymerisation, Blackwell Publishing, India, 2005 Search PubMed
- R. Wang, J. Ma, X. Zhou, Z. Wang, H. Kang, L. Zhang, K. C. Hua and J. Kulig, Design and Preparation of a Novel Cross-Linkable, High Molecular Weight and Bio-Based Elastomer by Emulsion Polymerization, Macromolecules, 2012, 45, 6830 CrossRef CAS
- A. J. Johanson, F. L. McKennon and F. L. Goldblatt, Emulsion Polymerisation of Myrcene, Ind. Eng. Chem., 1948, 40, 500 CrossRef CAS
- D. K. Kotnees, S. Gupta, B. B. Sharma, H. Tsou Andy and A. K. Bhowmick, Probing the Viscoelastic Properties of Brominated Isobutylene-co-p-Methylstyrene Rubber/Tackifier Blends Using a Rubber Process Analyzer, Polym. Eng. Sci., 2008, 48, 2400 Search PubMed
- A. L. Gergely, Ph.D. thesis, University of Akron, 2014
- R. Newmark and R. Majumdar, 13C-NMR Spectra of cis-Polymyrcene and cis-Polyfarnesene, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 71 CrossRef CAS
- F. Stoffelbach, L. Tibiletti, J. Rieger and B. Charleux, Surfactant-Free, Controlled/Living Radical Emulsion Polymerization in Batch Conditions Using a Low Molar Mass, Surface-Active Reversible Addition-Fragmentation Chain-Transfer (RAFT) Agent, Macromolecules, 2008, 41, 7850 CrossRef CAS
- S. V. Kostjuk, S. Ouardad, F. Peruch, A. Deffieux, C. Absalon, J. E. Puskas and F. Ganachaud, Carbocationic Polymerization of Isoprene Co-initiated by B(C6F5)3: An Alternative Route Toward Natural Rubber Polymer Analogues?, Macromolecules, 2011, 44, 1372 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision, Gaussian Inc., Wallingford, CT, 2010 Search PubMed
- A. Gula, Z. Akhterb, R. Qureshib and S. Bhattia Arshad, Conducting Poly(azomethine)ester: Synthesis, Characterization and Insight into electronic properties using DFT calculations, RSC Adv., 2014, 4, 22094 RSC
- R. C. Michael, Glass Transition in Rubbery Materials, ACS Rubber Chemistry and Technology, 2012, 85, 313 CrossRef
- M. P. Luda, M. Guaita and O. Chiantore, Thermal Drgradation of Polybutadiene, 2 Overall Thermal Behaviour of Polymers with Different Microstructure, Macromol. Chem. Phys., 1992, 193, 113 CrossRef CAS
- S. Sadhu and A. K. Bhowmick, Unique Rheological Behaviour of Rubber Based Nanocomposites, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1854 CrossRef CAS
- A. K. Bhowmick, P. P. De and A. K. Bhattacharyya, Adhesive Tack and Green Strength of EPDM Rubber, Polym. Eng. Sci., 1987, 27, 1195 CAS
Footnote |
| † Electronic supplementary information (ESI) available: Wide angle XRD analysis, particle morphology of the latex emulsion. See DOI: 10.1039/c4ra09475a |
| This journal is © The Royal Society of Chemistry 2014 |