
Synthesis and characterization of well-defined PAA–PEG multi-responsive hydrogels by ATRP and click chemistry†
Abstract
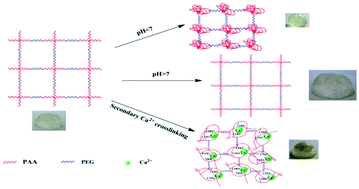
Multi-responsive poly(acrylic acid)–poly(ethylene glycol) (PAA–PEG) hydrogels with well-defined crosslinking structures were synthesized using atom transfer radical polymerization (ATRP) and copper-catalyzed 1,3-dipolar azide-alkyne cycloaddition (CuAAC) techniques. The well-defined PAA–PEG hydrogels with different degrees of crosslinking were produced from controlling the molecular weight of the PAA and PEG chains. The prepared multi-responsive hydrogels exhibit regular physical and mechanical properties by adjusting the pH and Ca2+ ion secondary crosslinking. With increasing pH, the swelling ratio of the well-defined multi-responsive PAA–PEG hydrogels increased remarkably. Furthermore, the well-defined PAA–PEG hydrogels with Ca2+ secondary crosslinking possessed a significantly higher crosslinking density as reflected by the lower swelling ratio, higher storage modulus, higher electrical conductivity and thermal stability. An in vitro cell viability assay also indicated that well-defined multi-responsive PAA–PEG hydrogels are biocompatible and have potential for implantable biomaterials.
 Please wait while we load your content...
Please wait while we load your content...

