
Superactivity of α-chymotrypsin with biological buffers, TRIS, TES, TAPS, and TAPSO in aqueous solutions†
Bhupender S. Guptaa,
Mohamed Tahab and
Ming-Jer Lee*a
aDepartment of Chemical Engineering, National Taiwan University of Science and Technology, 43 Keelung Road, Section 4, Taipei 106-07, Taiwan. E-mail: mjlee@mail.ntust.edu.tw
bCICECO, Departamento de Química, Universidade de Aveiro, 3810-193 Aveiro, Portugal
First published on 8th October 2014
Abstract
Biological buffers are always considered as non-toxic, biocompatible and green compounds. Therefore, we analyzed the catalytic activity of a commercial enzyme α-chymotrypsin (α-CT) in aqueous solutions of some common biological buffers (TRIS, TES, TAPS, and TAPSO) at pH 8 and T = 25 °C. It is found that the increase of the buffer concentration enhanced the catalytic activity of enzyme α-CT, and the tendency follows the order of TRIS > TES > TAPS > TAPSO. Especially in the presence of TRIS, the catalytic activity enhanced 5.5 fold.
A synthetic process using an enzyme as a catalyst is always considered as a green process. The catalyst efficiently increases the selectivity of the reaction system and significantly decreases the required processing time. Since the application of enzyme as a catalyst makes the process not only more economical but also faster. The process involving enzyme catalysis is highly concerned in the industries. However, industrial applications of enzymes are limited by their substantial degree of structural variability in folded state. Therefore, the knowledge of structure and function of enzymes in co-solvent are fundamentally important for understanding their metabolic role and for designing new synthetic applications. Unfortunately, enzymes are highly complex and extremely sensitive and they undergo reversible or irreversible conformational changes in response to any internal or external stress. It is recognized that the enzymes are biologically active only in their native conformation. Any diversion from native conformation usually leads to the loss in bioactivity of enzymes or proteins. Therefore, a co-solvent that can stabilize the structure of enzyme or enhance its activity is highly desirable in order to convert many existing non-green synthetic process into the green and sustainable process. Researchers have recognized the importance of the active conformation of biomolecules, especially in aqueous solution, and have developed many co-solvents to stabilize the structure of proteins.1–8 For example; polyhydric alcohol and sugars are recognized as strong stabilizers for proteins.9–13 Lately, ionic liquids emerged as a new class of biocompatible solvents for biomolecules.14–18
However, all the stabilizing or enhancing activity solutions so far reported for biomolecules cannot avoid the buffer from being a part of the system, since enzymes are only found to be active under their respective pH range. Thus, it is impossible to think enzyme without buffer. An ideal buffer is considered as non-toxic, biocompatible and environmental friendly. Buffer molecules are capable of maintaining the stable pH range for biomolecules to work efficiently. Extensive researches have been done on biomolecules in the presence of buffers, but very few efforts have been made to drive the mechanism about enzyme–buffer interactions or the consequences of such interactions on the structure and the applications of enzyme. Therefore, in this study we have investigated the effects of some important biological buffers, tris(hydroxymethyl)aminomethane (TRIS), N-[tris(hydroxymethyl)methyl]-2-aminoethane-sulfonic-acid (TES), N-[tris(hydroxylmethyl)methyl]-3-aminopropanesulfonic acid (TAPS), and N-[tris(hydroxymethyl)methyl]-3-amino-2-hydroxypropanesulfonic acid (TAPSO) on the activity of α-CT to catalyze the hydrolysis of p-nitrophenylacetate (PNPA). The hydrolysis of PNPA catalyzed by CT is well known and it has attracted great attention from enzymologists.19–22 The selected α-CT is a serine protease enzyme that is widely distributed in nature and performs a variety of major functions.23 α-CT is also commonly used in pharmaceutical, food, and health care products. It specializes in catalyzing many biochemical reactions to be absorbed on the surface of some substances and to be bound with other molecules and to form molecular aggregates.24
The activities of α-CT in aqueous solution of buffer were measured by the same method as described elsewhere.25,26 The activities of α-CT in 0.05 M, 0.5 M, and 1.0 M TRIS, TES, TAPS and in 0.05 M, 0.5 M, and 0.7 M TAPSO were examined by estimating the appearance of the p-nitrophenoxide (PNP−) at 400 nm, at pH 8 and at 25 °C with the help of UV-Visible spectrophotometer (JASCO, V-550). The enzyme activity was calculated by using the following equation:
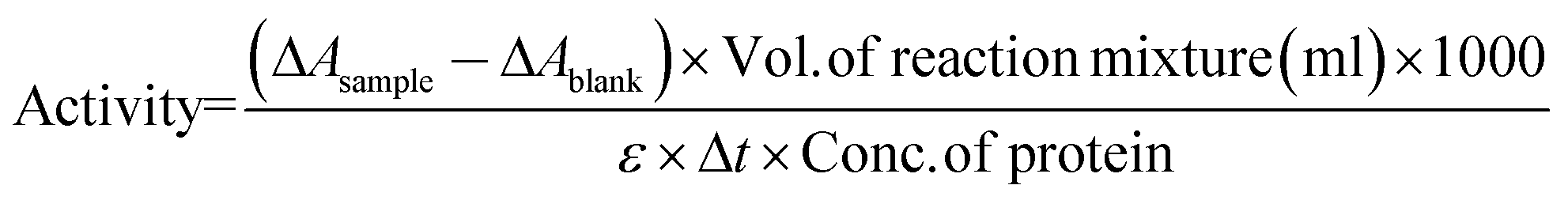
 | ||
| Fig. 1 (a and b). The relative activity of α-CT in 0.5 M, and 1.0 M TES, TAPS, and TAPSO buffer solutions at pH 8.0 and temperature 25 °C, after 3 minutes: (a) at 0.5 M, and (b) at 1.0 M. | ||
To understand the mechanism of α-CT-buffer interaction and to acquire the information about the conformational and the structural changes in enzyme α-CT caused by buffer molecules, we have performed spectroscopic measurements such as UV-Visible and fluorescence for the aqueous solution of α-CT in 0.05 M, 0.5 M, 0.7 M, and 1.0 M TRIS, TES, TAPS, and TAPSO buffer at 25 °C and pH = 8. Chervenka30 has reported that the UV-Visible spectra of α-CT in aqueous solution exhibits two absorption peaks at 200–220 nm and 260–300 nm, respectively. The first peak in the lower wavelength region (200–220 nm) reflects the backbone framework of the protein and it corresponds to the π → π* transition.31 The second peak in the higher wavelength region (260–300 nm) relates to the aromatic amino acids such as tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe) and the result of the n → π* transition.32 It can be seen from Fig. 2 that the first peak of α-CT in 0.05 M TRIS has a maximum at 218 nm. This peak shifts toward a higher wavelength and becomes less intense with the increasing TRIS concentration. For example, the peak has a maximum at 223 nm in 0.5 M TRIS, and at 224 nm in 1.0 M TRIS. The ESI Fig. 1S(a–c)† explicitly elucidates that the trend of the red shift in spectra of α-CT with the increasing buffer concentration is maintained with each studied buffer. Glazer and Smith33 reported that the amide group of the protein exposed to the aqueous environment undergoes the π–π* transition on ultraviolet irradiation. Later, Steinhardt34 explained that the red shift in the peak of protein in the lower wavelength region may be caused by the change in polarity of the solvent, especially in the vicinity of amide moiety. These buffers (TRIS, TES, TAPS, and TAPSO) are highly polar compounds and are known to have high solubility in water.29 It is probable that the increased polarity of the solvent with the increased respective buffer concentration induced the shielding of the peptide group from the aqueous environments and lowered the energy of the π and π* electron clouds assisting the low-energy π–π* transition. This lower energy π–π* transition appears as the bathochromic shift in the peak of α-CT in the lower wavelength region. This explains why the red shifts increase with the increasing respective buffers concentration. The peak of α-CT corresponding to the aromatic amino acid residues (Trp, Tyr, and Phe) remains unchanged irrespective of the type of buffer and concentration. This peak appears to have a maximum at 281 nm in each of the studied buffer condition. The small change in the intensity of this peak of α-CT was observed from the samples containing TES, TAPS, or TAPSO with various concentrations. The observed minor changes may be the results of the change in micro-environment around aromatic amino acid residues due to the conformational change in the protein structure.
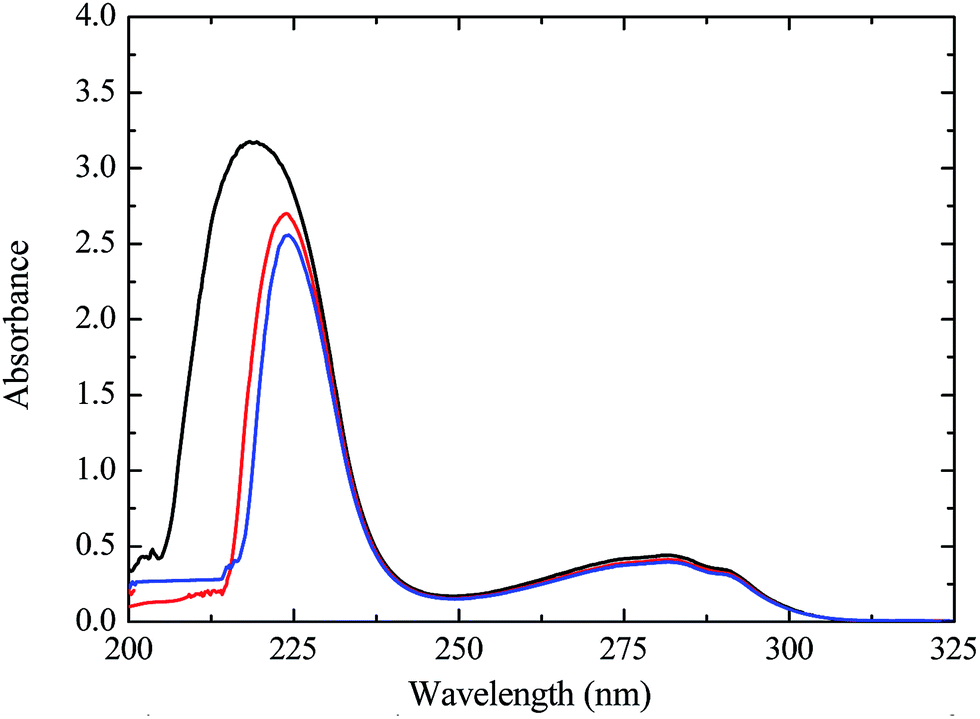 | ||
| Fig. 2 The UV-Visible measurements for α-CT in 0.05 M, 0.5 M, and 1.0 M TRIS at 25 °C and pH 8.0. Solid black line 0.05 M, solid red line 0.5 M, and solid blue line 1.0 M. | ||
Since the fluorescence intensity of fluorophore molecule is highly sensitive to the molecular environment,35 therefore, fluorescence technique serves as an efficient tool in the investigation of molecular environment in the vicinity of chromophore molecule. In case of proteins, the aromatic amino acid residues, such as tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe) are responsible fluorophore molecule. However, it was observed that, when enzyme is excited at or above 295 nm, the contributions from Tyr or Phe residues became negligible.36 Consequently, the emission spectra of α-CT at 295 nm mainly appear due to the Trp residues. The enzyme under investigation (α-CT) knows to have eight Trp amino acid residues. Therefore, to investigate the structural changes of the enzyme with solvent composition, we have measured the fluorescence spectra of α-CT in different concentrations (0.05 M, 0.5 M, 0.7 M, and 1.0 M) of aqueous buffer solutions (pH = 8) at excitation wavelength of 295 nm. The results of measurement for TRIS presented in Fig. 3 and for other buffers (TES, TAPS, and TAPSO) are compiled in ESI Fig. 2S(a–c).† The crystallographic study on the atomic structure of native α-CT36 revealed that most of the tryptophan residues in α-CT such as Trp27, Trp29, Trp51, Trp141, Trp172 and Trp215 are less exposed to the external solvent. While, two tryptophan residues (Trp207 and Trp237), are easily accessible to the external solvent. Based on these structural features of tryptophan residues in α-CT, it was assumed that the effect of solvent on the emission spectra of the α-CT attributed to the change in environmental polarity of the tryptophan residue. This change in environmental polarity of tryptophan residues may be caused either by solvent polarity or the structural change in enzyme.37–39
 | ||
| Fig. 3 The emission fluorescence spectra for α-CT in 0.05 M, 0.5 M, and 1.0 M TRIS at excited wavelength of 295 nm, 25 °C, and pH 8.0: (■), 0.05M; (●), 0.5 M: (▲), 1.0 M. | ||
A close analysis of the fluorescence spectra of α-CT in Fig. 3 and 2S(a–c)† reveals that with the increase of buffer concentration, the fluorescence peak shifts towards lower wavelength with minor increase in intensity. The effect is very small in case of TAPS or TAPSO, but is observable in TRIS or TES. This finding supports that the Trp environment is more perturbed in the presence of TRIS and TES in comparison with TAPS and TAPSO. It is well known that the increase in hydrophilic environment around Trp residues may cause the shifting in the emission peak towards higher wavelength and the shifting in the emission peak toward lower wavelength corresponds to increasing in the hydrophobic microenvironment around Trp residues. Therefore, the Trp residue shielded from the aqueous environment undergoes blue shift with high quantum yield. It might be possible that this shielding effect may provide a more stable and compact native structure to the protein in the solvent medium. This expectation is supported by our previous report,40 in which we have observed the hydrophobic collapse of thermo responsive polymer poly(N-isopropylacrylamide) (PNIPAM) in aqueous solutions of buffers, 2-(N-morpholino)ethanesulfonic acid (MES), 3-(N-morpholino)propanesulfonic acid (MOPS), and 3-morpholino-2-hydroxypropanesulfonic acid (MOPSO) at very low temperature in comparison with PNIPAM in water. PNIPAM is an isomer of polyleucine, and is known as a model compound for protein. The effect of these buffers on LCST of PNIPAM found to increase with increase of respective buffer concentration. On the basis of series of studies, we have found that with the increase of buffer concentration, the buffer–water interactions became dominant over polymer–water interactions that drive the hydrophobic collapse of the polymer at comparatively low temperature. It has been known that the buffers (TRIS, TES, TAPS, and TAPSO) are polar compounds and are highly soluble in water.29 It suggests that, in the present case, these buffers molecules may withdrawing the hydration layer around enzyme and thus providing more compact structure to the protein with native conformation, especially at higher concentrations (0.5 M, 0.7 M, and 1.0 M) of respective buffer. Probably this compactness in the structure causes the microenvironment around the Trp residues comparatively hydrophobic. These results indicate that with the increase of buffer concentrations, solvent polarity increases, which provides the comparatively stable and compact native conformation to the protein. This might be the reason why the catalytic activity of α-CT increased with the increase of the respective buffer concentration. Our expectations are in agreement with those provided by Dovaska-Taran et al.,41 who explained that the blue shift in the emission spectra of α-CT is attached with anthraniloyl fluorophore with the increasing surfactant concentration.
To further investigate the possible binding sites of the studied buffers (TRIS, TES, TAPS, and TAPSO) in enzyme α-CT, we have performed the molecular docking study between enzyme α-CT and biological buffers TRIS, TES, TAPS, and TAPSO with the Auto-dock–vina 1.5.4 docking program.42 The results of molecular docking are presented in the Fig. 4 for TRIS and in the ESI Fig. 3S(a–c)† for TES, TAPS, and TAPSO, respectively. These figures show that all these buffers do not have any direct interaction with the catalytic site (His 57, Asp 102 and Ser 195) of α-CT. The number of hydrogen bonds formed by TRIS, TES, TAPS, and TAPSO buffers with adjacent amino acid residues are 10, 8, 5, and 9, respectively. The results show that the main interaction between the investigated buffer and enzyme α-CT is electrostatic. However, the α-CT-buffer interaction is not unique with each buffer. The different buffers have been observed to interact with different amino acid residues of α-CT.
 | ||
| Fig. 4 The hydrogen bond interactions of TRIS with α-CT. | ||
Dzhafarov43 has investigated the interactions of fatty acid with the albumin protein. The major effect of the binding of fatty acid with albumin protein is reflected as the change in the shape with an increase in the stability of the protein structure. Perhaps, the noticed bindings between the studied buffer and the globular enzyme α-CT change the shape of the enzyme α-CT to the more active conformation in the same manner as done by fatty acid. Kotila and Valkonen44 have reported that TRIS acted as a polydentate ligand and forms chelates with transition metal ion by using amino group and one or two hydroxyl groups. It can be seen clearly from Fig. 4 that the amino group and all the hydroxyl groups of TRIS participating in hydrogen bonding with different amino acid residues of α-CT. In addition, it appears to interact with maximum number of amino acid residues of α-CT. In contrast, TAPS buffer is found to interact with the least number of amino acid residues and forming minimum number of hydrogen bonds with adjacent amino acids (Fig. 3S(b)†). This finding explains why TRIS appears the strongest and TAPS appears the weakest buffer in enhancing the activity of α-CT. TES buffer is less hydrophobic and more polar than TAPS and TAPSO buffer. Thus, the observed electrostatic interactions, including hydrogen bonding, in TES system are stronger than those in TAPS or TAPSO system. Since these interactions are stabilizing the native conformation of the enzymes, this might be the reason, why TES buffer is more promising in enhancing catalytic activity of α-CT in comparison with TAPS or TAPSO. TAPS and TAPSO buffers are structurally similar, except the presence of an extra hydroxyl group (–OH) between the amino and the sulfonic group in TAPSO. It is interesting to see that this extra hydroxyl group of TAPSO involved in the hydrogen bonding with Gln34 and Gly38 (Fig. 3S(c)†). It might be the reason for the enhanced catalytic activity of the α-CT in TAPSO over TAPS. Thus, the outcomes from molecular docking are in agreement with the experimental findings. From this series of systematically studies, it is revealed that the investigated buffers are not only suitable for maintaining the required pH value in aqueous solutions but also can be used to enhance the activity of enzymes.
Conclusions
The catalytic activity of enzyme α-CT was analyzed in the presence of the commonly used biological buffers: TRIS, TES, TAPS, and TAPSO. Interestingly, all the studied buffers were found to enhance the activity of enzyme and the activity enhancement increases with increasing buffer concentration. Since the enzymatic catalyzed processes are widely used and are highly important from industrial point of view, the current study can help in the improvement of such process in a more economical way. In addition, we investigated the mechanism of the interaction of these buffers with α-CT. From the series of carefully performed analysis on buffer-α-CT systems, the results reveal that the buffer molecules, at higher concentrations, provide more compact conformation to the enzyme. The main interaction between buffer and α-CT is electrostatic. Certainly, such information can help in the better understanding of the process based on the buffer–enzyme systems. Therefore, employing buffer as an enzyme stabilizer or an activity enhancer can be an advantageous alternative over using the traditional volatile organic compounds and the expensive modern ionic liquids.Acknowledgements
The authors gratefully acknowledge the financial support from the National Science Council, Taiwan, through Grant no. NSC102-2811-E-011-014 and NSC 102-2221-E011-137-MY3.Notes and references
- E. V. Kudryashova, A. K. Gladilin, A. V. Vakurov, F. Heizt, A. V. Levashov and V. V. Mozhaev, Biotechnol. Bioeng., 1997, 55, 267–277 CrossRef CAS.
- V. V. Levitsky, P. Lozano and J. L. Iborra, Biotechnol. Lett., 1999, 21, 595–599 CrossRef CAS.
- L. M. Simon, M. Kotormán, G. Garab and I. Laczkó, Biochem. Biophys. Res. Commun., 2001, 280, 1367–1371 CrossRef CAS PubMed.
- B. S. Gupta, M. Taha and M. J. Lee, Process Biochem., 2013, 48, 1686–1696 CrossRef CAS PubMed.
- A. A. Vinogradov, E. V. Kudryashova, V. Y. Grinberg, N. V. Grinberg, T. V. Burova and A. V. Levashov, Protein Eng., 2001, 14, 683–690 CrossRef CAS PubMed.
- A. Kumar and P. Venkatesu, Chem. Rev., 2012, 112, 4283–4307 CrossRef CAS PubMed.
- A. Kumar, P. Attri and P. Venkatesu, Thermochim. Acta, 2012, 536, 55–62 CrossRef CAS PubMed.
- P. Attri, P. Venkatesu and M. J. Lee, J. Phys. Chem. B, 2010, 114, 1471–1478 CrossRef CAS PubMed.
- J. Wyman and S. Gill, Binding and linkage: functional chemistry of biological molecules, University science books, California, Mill Valley, 1990 Search PubMed.
- S. N. Timasheff and T. Arakawa, Stability of protein structure by solvents, in Creighton TE editors. Protein Structure: A Practical Approach, Oxford University Press, Oxford, 1989 Search PubMed.
- S. N. Timasheff, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 67–97 CrossRef CAS PubMed.
- S. N. Timasheff, Adv. Protein Chem., 1998, 51, 356–432 CrossRef.
- M. T. Record, W. Zhang and C. Anderson, Adv. Protein Chem., 1998, 51, 282–355 CrossRef.
- K. Rawat and H. B. Bohidar, J. Phys. Chem. B, 2012, 116, 11065–11074 CrossRef CAS PubMed.
- D. H. Dagade, K. R. Madkar, S. P. Shinde and S. S. Barge, J. Phys. Chem. B, 2013, 117, 1031–1043 CrossRef CAS PubMed.
- M. Bekhouche, L. J. Blum and B. Doumeche, J. Phys. Chem. B, 2012, 116, 413–423 CrossRef CAS PubMed.
- Y. Akdogan, M. J. N. Junk and D. Hinderberger, Biomacromolecules, 2011, 12, 1072–1079 CrossRef CAS PubMed.
- C. A. Summers and R. A. Flowers II, Prot. Sci., 2000, 9, 2001–2008 CrossRef CAS PubMed.
- N. Spetri, F. Alfani, M. Cantarella, F. D. Amico, R. Germani and G. Savelli, J. Mol. Catal. B: Enzym., 1999, 6, 99–110 CrossRef.
- P. Viparelli, F. Alfani and M. Cantarella, J. Mol. Catal. B: Enzym., 2001, 15, 1–8 CrossRef CAS.
- P. Viparelli, F. Alfani and M. Cantarella, J. Mol. Catal. B: Enzym., 2003, 21, 174–187 CrossRef.
- P. Viparelli, F. Alfani and M. Cantarella, Biochem. J., 1999, 344, 765–773 CrossRef CAS.
- P. Attri, P. Venkatesu and A. Kumar, Phys. Chem. Chem. Phys., 2011, 13, 2788–2796 RSC.
- D. G. Dalgleish, in Emulsions and emulsion stability, ed. J. Sjoblom, New York, Marcel Dekker, 1996, ch. 5 Search PubMed.
- P. Attri, P. Venkatesu and M. J. Lee, J. Phys. Chem. B, 2010, 114, 1471–1478 CrossRef CAS PubMed.
- B. F. Erlanger, N. Kokowsky and W. Cohen, Arch. Biochem. Biophys., 1961, 95, 271–278 CrossRef CAS.
- T. De-Diego, P. Lozano, S. Gmouth, M. Vaultier and J. L. Iborra, Biotechnol. Bioeng., 2004, 75, 916–924 CrossRef PubMed.
- G. R. Castro, Enzyme Microb. Technol., 2000, 27, 143–150 CrossRef CAS.
- M. Taha and M. J. Lee, Phys. Chem. Chem. Phys., 2010, 12, 12840–12850 RSC.
- C. H. Chervenka, Chlorure De Sodium Et Autolyse De Trpsine., 1959, 31, 85–95 CAS.
- Q. Yan, J. Liang and H. Han, J. Phys. Chem. B, 2009, 113, 10454–10458 CrossRef PubMed.
- X. C. Zhao, R. T. Liu, Z. X. Chi, Y. Teng and P. F. Qin, J. Phys. Chem. B, 2010, 114, 5625–5631 CrossRef CAS PubMed.
- A. N. Glazer and E. L. Smith, J. Biol. Chem., 1961, 236, 2942–2947 CAS.
- H. Polet and J. Steinhardt, Biochemistry, 1968, 7, 1348–1346 CrossRef CAS.
- N. C. Stellwagen, A. Bossi, C. Gelfi and P. G. Righetti, Anal. Biochem., 2000, 287, 167–175 CrossRef CAS PubMed.
- P. Attri and P. Venkatesu, Process Biochem., 2013, 48, 462–470 CrossRef CAS PubMed.
- M. Moller and A. Denicola, Biochem. Mol. Biol. Edu., 2002, 30, 175–178 CrossRef CAS.
- N. Rezaei-Ghaleh, A. Ebrahim-Habibi, A. A. Moosavi-Movahedi and M. Nemat-Gorgani, Biochem. Biophys., 2007, 457, 160–169 CrossRef CAS PubMed.
- I. Pallares, J. Vendrell, F. X. Aviles and S. Ventura, J. Mol. Biol., 2004, 342, 321–331 CrossRef CAS PubMed.
- M. Taha, B. S. Gupta, I. Khoiroh and M. J. Lee, Macromolecules, 2011, 21, 8575–8589 CrossRef.
- V. N. Dorovska-Taran, C. Veeger and A. J. W. G. Visser, Eur. J. Biochem., 1993, 211, 47–55 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- E. S. Dzhafarov, Mol. Biol., 1992, 26, 168–172 CAS.
- S. Kotila and J. Valkonen, Acta Chem. Scand., 1994, 48, 312–318 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09434d |
| This journal is © The Royal Society of Chemistry 2014 |