
Self-assembly of mucoadhesive nanofibers†
Abstract
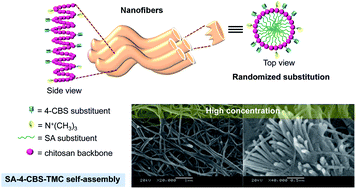
Here, we present an easy and stable one-step procedure to fabricate self-assembled nanofibers from modified chitosan. To obtain self-assembled, well-ordered nanofibers, we designed and synthesized stearic acid-4-carboxybenzenesulfonamide-N-trimethylchitosan by only re-dispersing the compound in distilled water at a concentration of 3.33 mg mL−1. The self-assembled nanofibers had a diameter of 112.23 ± 11.96 nm with a narrow width distribution obtained through the aromatic stacking of 4-carboxybenzenesulfonamide, the hydrophobic effect of the stearic acid and the hydrogen bonding of the chitosan backbone. The intercoronal interaction of –N+(CH3)3 provided an elongated axis of nanofibers. Furthermore, the ordered molecular organization of nanofibers led to high thermal stability and enhanced mucoadhesive properties compared to native chitosan, making the fabrication of these nanofibers a promising assembly method for drug delivery in acidic environments.
 Please wait while we load your content...
Please wait while we load your content...

