
The effect of epoxy–silicone copolymer content on the thermal and mechanical properties of cured epoxy resin modified with siloxane†
Yang Chen,
Cheng Zhou,
Jin Chang,
Huawei Zou* and
Mei Liang*
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Sichuan University, Chengdu 610065, China. E-mail: hwzou@163.com; liangmeiww@163.com; Fax: +86-28-85402465; Tel: +86-28-85408288
First published on 24th October 2014
Abstract
In this research, a bisphenol-A type epoxy resin (DGEBA) was modified with epoxy-block-silicone copolymers and hydroxyl-terminated silicone oligomers by physical blending and a chemical reaction, respectively. The chemical structure of the siloxane-bridged epoxy resin terminated by –OHs was characterized by Fourier transform infrared spectroscopy (FTIR), 1H-NMR and an epoxy equivalent weight (EEW) test. Both the samples showed better elongation at break and impact strength than neat resin. The TGA-FTIR results revealed that the residue of the modified epoxy resin at 600 °C increased with the increase in siloxane content, but the thermal stability was slightly reduced compared with that of the neat epoxy resin. Morphology studies indicate that the increase in izod notched impact strength is due to the suitable diameter of silicone phases because of the silicone toughening effect. Therefore, it is believed that the modified epoxy resin, with good toughness and high thermal residual weight, will have potential applications in anti-corrosion coatings and structure bonding materials.
1. Introduction
Epoxy resin has been widely used in many applications such as composite materials, coatings, structural adhesives and microelectronics1,2 due to its good mechanical properties, excellent chemical resistance, heat stability, excellent adhesion characteristics, and high electrical insulation. The major drawback of epoxy resin is the brittleness caused by its high degree of crosslinking. This inherent brittleness results in the poor impact strength of epoxy composites and the poor shear strength of epoxy-based adhesives.In light of this problem, researchers have incorporated carbon nanotubes,3,4 liquid crystalline polymers,5,6 rubber,7–9 and polyurethane10,11 to improve the toughness of cured epoxy resins. Of all these methods, the most successful one is the modification with reactive liquid rubbers, such as carboxyl-terminated butadiene acrylonitrile (CTBN),12,13 hydroxyl-terminated butadiene acrylonitrile (HTBN)14,15 and amine terminated butadiene acrylonitrile (ATBN).16,17 However, the presence of an unsaturated structure in liquid rubbers brings about thermal instability and low oxidation resistance into the epoxy matrix. Hence, such modified resins are not suitable for applications at high temperatures. In contrast, the modification of epoxy resin with organosilicone polymers can improve the toughness of cured epoxy without damaging its thermal resistance.18,19 However, polysiloxanes have poor compatibility with the epoxy resin due to their varied solubility parameters, which results in the formation of a finely phase-separated structure and a considerable sacrifice in the glass transition temperatures (Tg) in thermosets.20,21 Therefore, much attention has been paid to improving the compatibility between epoxy and silicone resins. For example, the introduction of an aramid–silicone block copolymer22,23 as a compatibilizer, preparation of silicon- and carbon-based block copolymers,24 and synthesis of polysiloxane copolymer which contains epoxide groups on the side chains19 have been proved to be effective. Studies for enhancing the toughness of epoxy resins evaluated in terms of the measurement of impact strength18,24–26 and critical stress intensity factor22,23,27 are reported. So far there have been few reports in the literature focused on the elongation at break aspects of silicone oligomer modified epoxy resins.
In this study, a commercial grade epoxy-block-silicone copolymer and hydroxyl-terminated silicone oligomers of different mass ratios were employed in epoxy resin by physical blending and a chemical reaction process, respectively. The mechanical and thermal properties and morphology of these modified epoxy resins were studied. The results indicate that the modified epoxy resin exhibits high elongation at break, good impact strength and advanced thermal stability compared to neat epoxy resin. Therefore, a high temperature filler could be introduced to the modified epoxy resins in order to prepare anti-corrosive coatings, electronic packaging and structural adhesive.
2. Experimental
2.1. Materials
Bisphenol-A type epoxy resins (DGEBA) with an epoxide equivalent weight of 185–192 g per eq. (n = 0.1) were purchased from Wuxi Resin Plant, China. Polypropylene oxide diglycidyl ether (DER732) with an average molecular weight of 640 g mol−1 was supplied by Dow Chemical Company. DER732 was used to reduce the viscosity in the curing process and to endow the system with excellent flexibility, elongation and impact properties. Hydroxyl-terminated silicone oligomer, with a brand name Z-6018, was obtained from Dow Corning, which is a mixture of phenyl, propyl silsesquioxanes (>60%) and a hydroxyl-terminated linear siloxane. The curing agent, polypropylene oxide Jeffamine (EC-301), was purchased from BASF Chemical Company (equivalent [H] weight: 61.0). Epoxy-block-siloxane copolymer (SH023) with an epoxy value of 0.02–0.07 mol per 100 g and a siloxane content of 70% was purchased from Hubei Zaoyang Company. Dibutyltindilaurate (DBTDL), which was used as a catalyst, was purchased from Chengdu Chemical Reagent Company, China. The chemical structures of DGEBA, Z-6018, DER732 and EC-301 are shown in Scheme 1. These materials were used as supplied without further purification.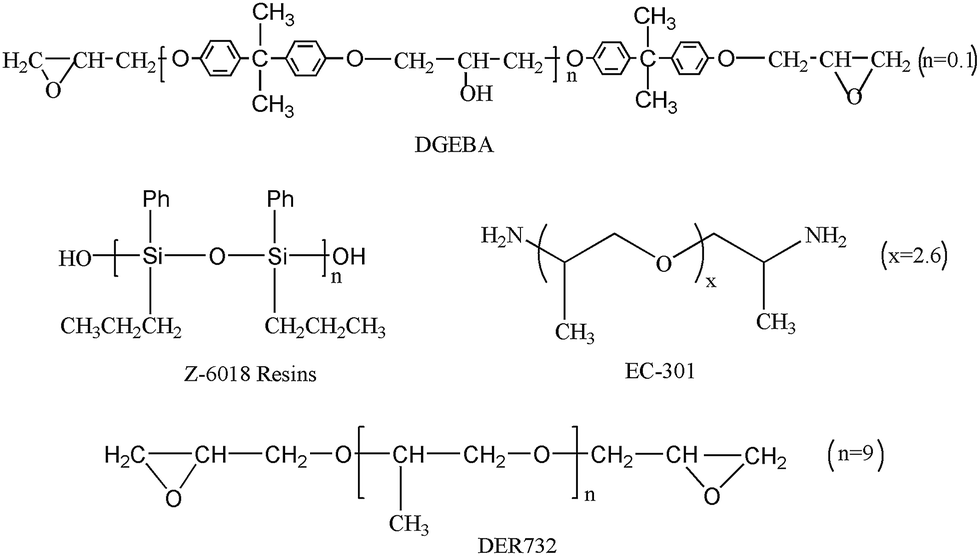 | ||
| Scheme 1 Chemical structures of DGEBA, Z-6018, DER732 and EC-301. | ||
2.2. Synthesis of hydroxyl-terminated silicone oligomer-bridged epoxy resins (PSG)
DGEBA was firstly fed into a 250 mL three-necked round-bottomed flask, which was equipped with a mechanical stirrer, a thermometer and a connection to a N2 cylinder. Epoxy resins were preheated to 120 °C in an oil bath and then a stoichiometric amount of Z-6018 was added and stirred under a nitrogen atmosphere. The compositions are shown in Table 1. Then a certain amount (0.5% of DGEBA resins) of DBTDL was added as a catalyst into the flask after Z-6018 had completely melted down. The polymerization was carried out at 120 °C for 2 h under a stirring rate of about 500 rpm. After the reaction was completed, colorless, transparent, viscous and homogeneous products were obtained (Scheme 2). The structure and the equivalent weight of these resultant products were confirmed by Fourier transform infrared spectrometry and the hydrochloric acid/acetone method, respectively.| Sample code | E-51 | PSG1 | PSG2 | PSG3 | PSG4 |
|---|---|---|---|---|---|
| Z-6018 (g) | 0 | 10 | 20 | 30 | 40 |
| DGEBA (g) | 80 | 70 | 60 | 50 | 40 |
 | ||
| Scheme 2 Synthesis of hydroxyl-terminated silicone oligomer-bridged epoxy resins (PSG). | ||
2.3. Curing of silicon modified epoxy resin
The PSDG copolymers were obtained by blending the reaction products of Section 2.2, which were used without further purification, and 25 wt% DER732 (based on the weight of the reaction products). Another kind of siloxane-type epoxy resin (PSHG) was prepared simply by blending alternating block polymers (SH023) with a stoichiometric amount of DGEBA and DER732. The formulations of SH023 and Z-6018 modified epoxy resins are shown in Table 2. Then a stoichiometric amount of the curing agent (EC-301) was added to PSDG and PSHG at room temperature. The equivalent ratio between the epoxy resin and the curing agent was based on the epoxide equivalent weight of epoxy resin and the amine equivalent weight (A.E.W.) of the curing agent. These blends were completely mixed by a mechanical stirrer and degassed in a vacuum oven to eliminate air bubbles. The bubble-free mixtures were poured into 4 mm-thick polytetrafluoroethylene (PTFE) molds which were preheated to 80 °C to prepare the specimens for mechanical tests. The materials were cured at 80 °C for 2.5 h and at 125 °C for 3 h in a convection oven.| Sample code | E-51 (g) | Z-6018 (g) | SH023 (g) | DER732 (g) | EC-301 (g) |
|---|---|---|---|---|---|
| a The total weight of the modified epoxy resins is 100 percent except curing agent. | |||||
| EG | 80.00 | 0 | 0 | 20.00 | 30.38 |
| PSDG1 | 70.00 | 10.00 | 0 | 20.00 | 24.52 |
| PSDG2 | 60.00 | 20.00 | 0 | 20.00 | 23.16 |
| PSDG3 | 50.00 | 30.00 | 0 | 20.00 | 20.08 |
| PSDG4 | 40.00 | 40.00 | 0 | 20.00 | 16.12 |
| PSHG1 | 70.00 | 0 | 10.00 | 20.00 | 27.38 |
| PSHG2 | 60.00 | 0 | 20.00 | 20.00 | 24.39 |
| PSHG3 | 50.00 | 0 | 30.00 | 20.00 | 21.40 |
| PSHG4 | 40.00 | 0 | 40.00 | 20.00 | 18.40 |
2.4. Characterization
A Fourier transform infrared (FTIR) spectrum was obtained by a FTIR spectrometer (Nicolet 570) using potassium bromide (KBr) pellets to investigate the structure of DGEBA, Z-6018 and PSG. The scans were collected in the range of 4000–400 cm−1 wavenumber at room temperature.Epoxide equivalent weights (EEWs) of the PSG systems were determined according to a GB 1677-81 standard. The analyses were performed in duplicate. The samples weighed 0.5–1.0 g, which were sufficient to determine the EEW values.
1H-NMR spectra were recorded on a DRX-400 spectrometer (Bruker Company, Germany) with CDCl3 solvent.
The tensile strength and breaking elongation of the cured specimens were conducted with the help of a Universal tensile instrument (INSTRON 5567) according to GB/T 2567-2008. The test was performed at a rate of 10 mm min−1 at room temperature.
The izod notched impact strength of the cured specimens was tested with an Izod Impact Tester (MTS Co.) according to GB/T 2567-2008. The size of the tested specimen was 4 mm × 10 mm × 80 mm. All mechanical property values were obtained by averaging the five experimental values.
The morphology of the impact fracture surfaces was observed using a scanning electron microscope (SEM:JSM-5900, JEOL Co., Ltd.) at an accelerating voltage of 10 kV. Prior to the examination, the fractured surfaces were coated with gold in order to enhance conductivity and prevent charging.
The thermal stability was studied by means of thermogravimetric analysis (TGA:Q500, TA Co, Ltd., USA) coupled with a FTIR spectrometer (Nicolet Magna 570, Nicolet Co, Ltd., USA). The sample weights ranged from 5–10 mg and were heated from 50 to 600 °C at a rate of 10 °C min in a dry nitrogen atmosphere. The flow rate of gases into the cell for the TGA/FTIR experiments was 100 mL min−1. The solid weight loss, together with other process variables such as temperature and gaseous species were detected using the FTIR spectrometer throughout the measurements.
3. Results and discussion
3.1. Characterization of hydroxyl-terminated silicone oligomer-bridged epoxy resins (PSG)
Fig. 1 shows the FTIR spectra of neat epoxy resins (DGEBA), hydroxyl-terminated silicone (Z-6018) and PSG1 (E51/Z-6018 = 70/10). For PSG1, the characteristic absorption peaks at 1438, 1247, 1133, 913 and 832 cm−1 were attributed to Si–C6H5, Si–CH3, Si–O–C and the terminal epoxy groups, respectively.28–33 Compared with Z-6018 and DGEBA, these peaks were ultimately nearly the same besides the diminished relative proportions of the peaks at 832 cm−1 and 913 cm−1. This indicates that only a small amount of the epoxy groups reacted with Si–O–H. The strong band in the range of 1035–1165 cm−1 becomes broader after the reaction with DGEBA, which was likely the result of the formation of a Si–O–C bond. However, the formation of the Si–O–C bond in PSG1 overlapped with the Si–O–Si band in the region of 1035–1165 cm−1 and 1200–1000 cm−1 of the C–O–C vibration. Moreover, the broad Si–O–H peak of Z-6018 (3140–3590 cm−1) changed to a narrow peak (3493 cm−1) of PSG1 attributed to the reaction between the Si–O–H and C–O–H of the epoxy resin (Scheme 2).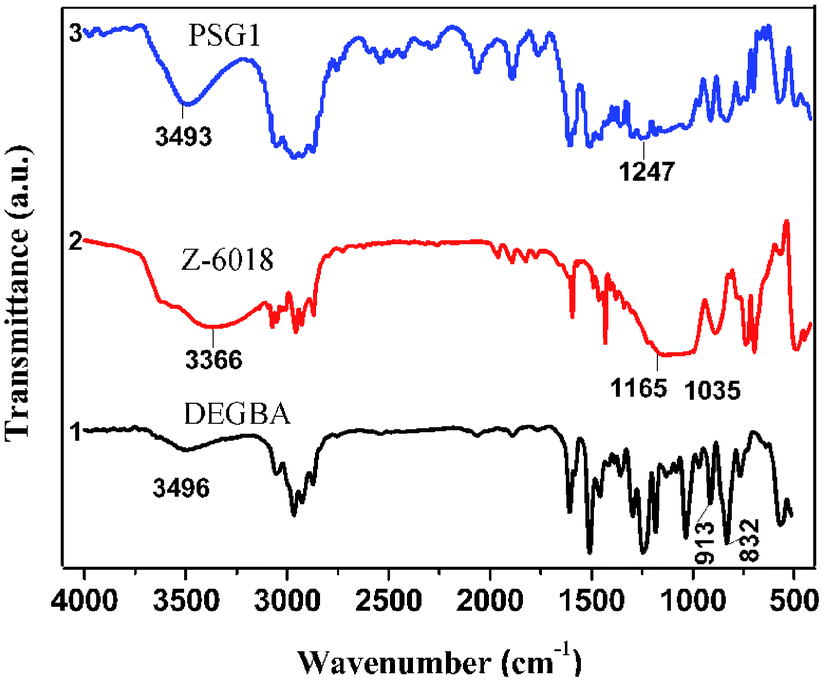 | ||
| Fig. 1 FTIR spectra of DGEBA, Z-6018 and PSG1. | ||
Pure epoxy resin (1H-NMR, CDCl3), δ (ppm): 7.14–6.8 (aromatic ring protons), 4.17 (–CH2–O-Ar), 3.33–3.30 (–CH, oxirane), 2.9–2.7 (–CH2, oxirane), 1.8–1.6 (–CH3). The 1H-NMR of PSG1 is shown in Fig. 2. The presence of characteristics peaks in the 1H-NMR spectra (δ, ppm, 2.2 (CH–O–Si)) of PSG1 is attributed to the reaction between Si–O–H and C–O–H of the epoxy resin and Z-6018 (Scheme 2).32 The results further support the formation of the PSG copolymer.
 | ||
| Fig. 2 1H-NMR spectra of the epoxy resin modified with silicone. | ||
The EEW values of the synthesized PSG copolymer are listed in Table 3. The calculated values, based on the weight ratios, agree well with the experimental data of the PSG copolymer. The opening rate of the epoxy ring remains almost unchanged. This further confirms that few epoxy groups are involve in the ring opening reaction. These results are consistent with the theoretical assumptions, 1H-NMR and FTIR results.
| Sample code | E-51 | PSG1 | PSG2 | PSG3 | PSG4 |
|---|---|---|---|---|---|
| a The EEW of E-51 is given by the product introduction of DGEBA.b The EEWs of PSG1, PSG2, PSG3, PSG4 are calculated based on the epoxy equivalent weight and the weight ratios of DGEBA. | |||||
| EEW (calculated value) (g mol−1) | 185–192a | 212b | 247b | 296b | 370b |
| EEW (experimental value) (g mol−1) | 185 | 238 | 255 | 304 | 378 |
| The rate of opening of the epoxy ring (%) | 0% | 11% | 3% | 2.6% | 2% |
3.2. Morphology of cured blends
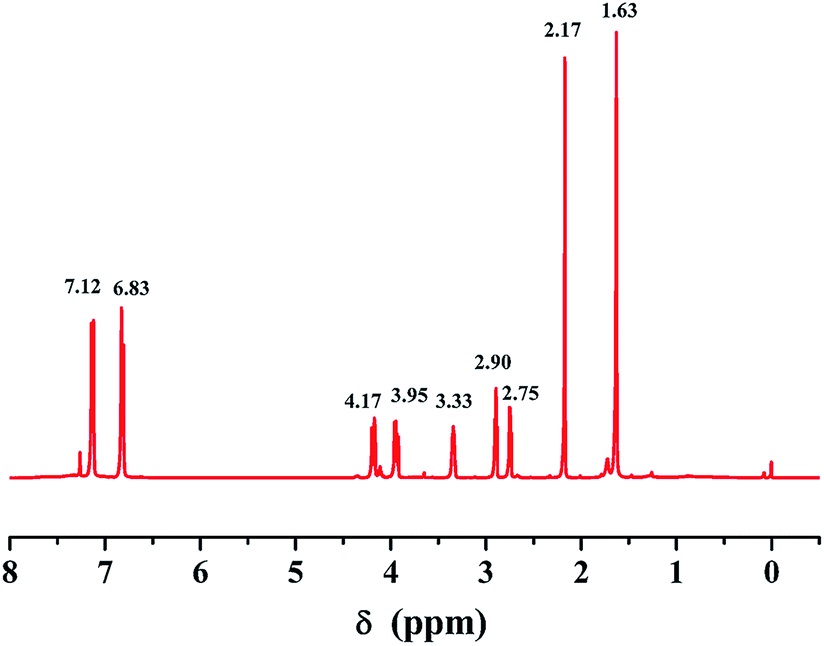
In Fig. 3(a), the EG (blank sample) exhibited a smooth, glassy microstructure without any plastic deformation. Fig. 3(b)–(d) show that the fine silicone phase was uniformly dispersed in the epoxy matrix by the addition of SH023. The average diameter of the dispersed phases of SH023 was 0.25 μm at the loading of 10 wt% and the size distribution was narrow. Moreover, the diameter of the silicone-dispersed phases increased to 0.81 μm with 40 wt% content of SH023. This means that SH023 shows good compatibility with epoxy resin. | ||
| Fig. 3 SEM micrographs of the fracture surfaces of epoxy resin modified with silicone: (a) EG, (b) PSHG1, (c) PSHG2, (d) PSHG4, (e) PSDG1, (f) PSDG2, (g) PSDG4. | ||
Fig. 3(e)–(g) show the morphology of the systems with Z-6018, which had silicone contents of about 10 wt%, 20 wt% and 40 wt%, respectively. A bi-phase separation structure was obviously observed through SEM. However, the SEM photographs show that better silicone phases could be uniformly dispersed in the epoxy matrix at the Z-6018 addition of 10 wt%, compared with the modified systems with 20 wt% and 40 wt% Z-6018. In particular, the diameter of the dispersed phases reached 2.8 μm in the system with 40 wt% Z-6018.
3.3. Mechanical properties
Fig. 4(a) and (b) show the tensile strength and elongation at break of the PSHG systems. The decreased tensile strengths with increasing SH023 content is ascribed to the low strength of SH023. Whereas, the breaking elongation of the systems increased almost seven times to 47% compared with that of epoxy resin when the SH023 content increase up to 40 wt%. The increased elongation at break can be attributed to the good compatibility between the epoxy-block-silicone copolymers and epoxy resin. The siloxane chain shows better deformability, while for the pure epoxy resins, the cross-linked network with poor chain mobility leads to low elongation at break.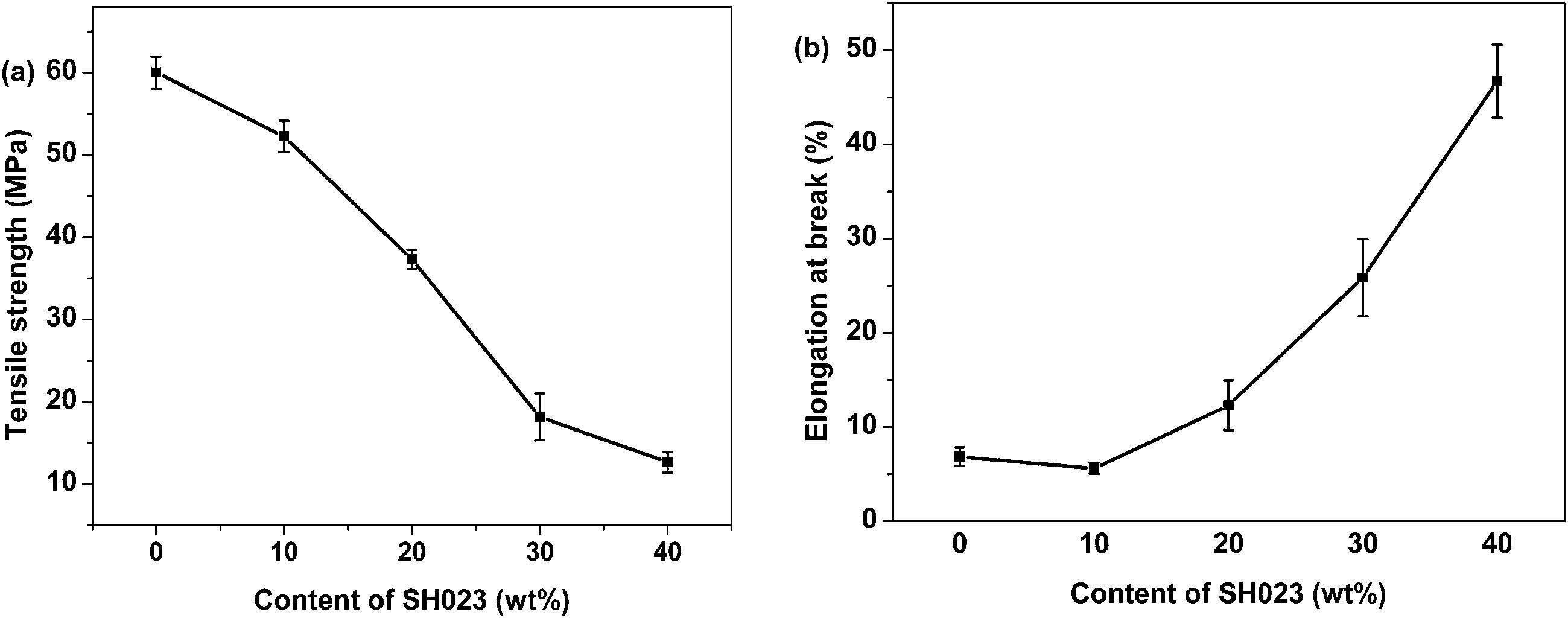 | ||
| Fig. 4 Tensile strength (a) and elongation at break (b) of the PSHG systems as a function of SH023 content. | ||
The values of tensile strength and elongation at break of the PSDG systems are illustrated in Fig. 5(a) and (b), respectively. The decline in tensile strength with an increase of Z-6018 is due to the presence of flexible siloxane linkages, free rotation of the Si–O–Si bonds, and weak interface boundaries between the siloxane and epoxy resin. The elongation at break showed few fluctuations until the Z-6018 content reached 40 wt% of the modified epoxy resins. Due to the bad compatibility between epoxy and silicone resins in PSDG systems, when a small amount of silicone is added, the –Si–O–Si– chains are not enough to change the network of epoxy resin, and therefore, the elongation at break remains unchanged.
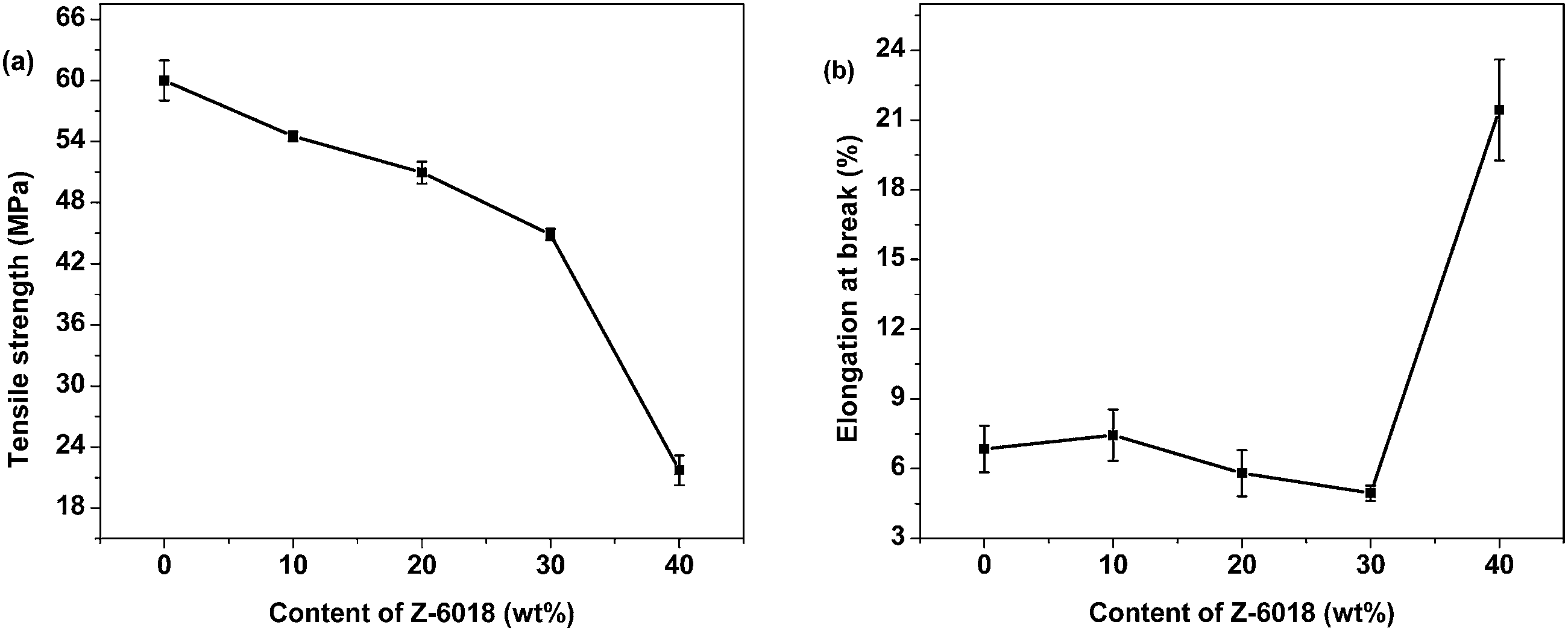 | ||
| Fig. 5 Tensile strength (a) and elongation at break (b) of the PSDG systems as a function of Z-6018 content. | ||
Fig. 6 shows the impact strength of the PSHG systems as a function of SH023 content. As observed, the impact strength values of the systems are significantly raised with increasing SH023 content. When the content of SH023 reaches 40 wt%, the impact strength of the systems increased nearly three times higher than that of the pure epoxy resins. This result can be interpreted in terms of increased energy dissipation induced by addition of a siloxane segment into the epoxy network.34,35 Siloxane chains act as obstacles in the crack path, and more energy is required for crack propagation.
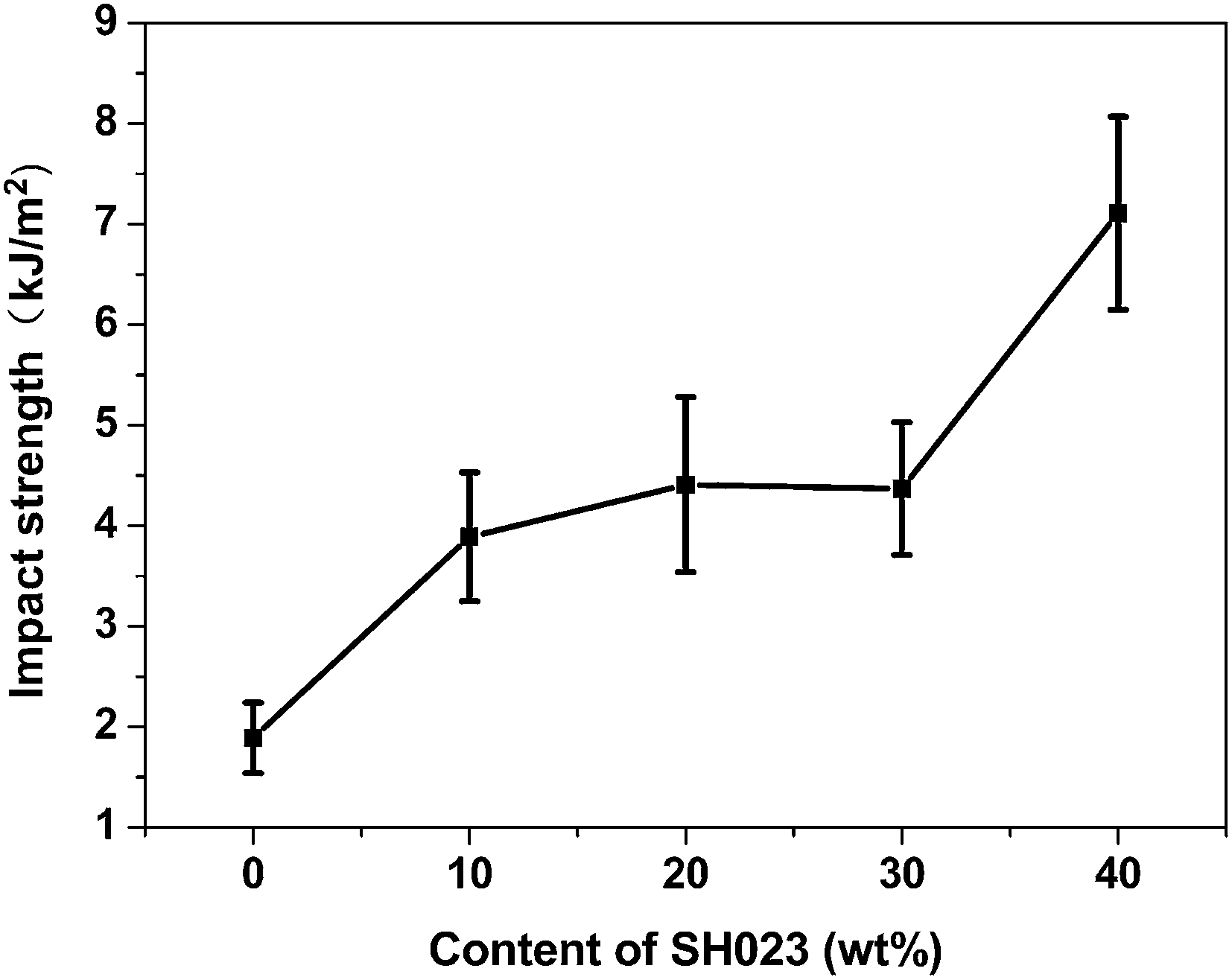 | ||
| Fig. 6 Impact strength of the PSHG systems as a function of SH023 content. | ||
Fig. 7 shows the impact strength values of the PSDG systems as a function of Z-6018 content. It was found that the impact strength of the modified systems has a maximum value when the Z-6018 content is 10 wt%. It can be explained that Z-6018 (10 wt%) has a good compatibility with epoxy resin. The silicone segment of Z-6018 is flexible enough to enhance the impact strength by increasing energy dissipation. When the Z-6018 content increased to more than 10 wt%, the compatibility of the epoxy resins and Z-6018 decreased. In addition, the diameter of the silicone phases became larger, thus resulting in poorer impact strength of the blends.
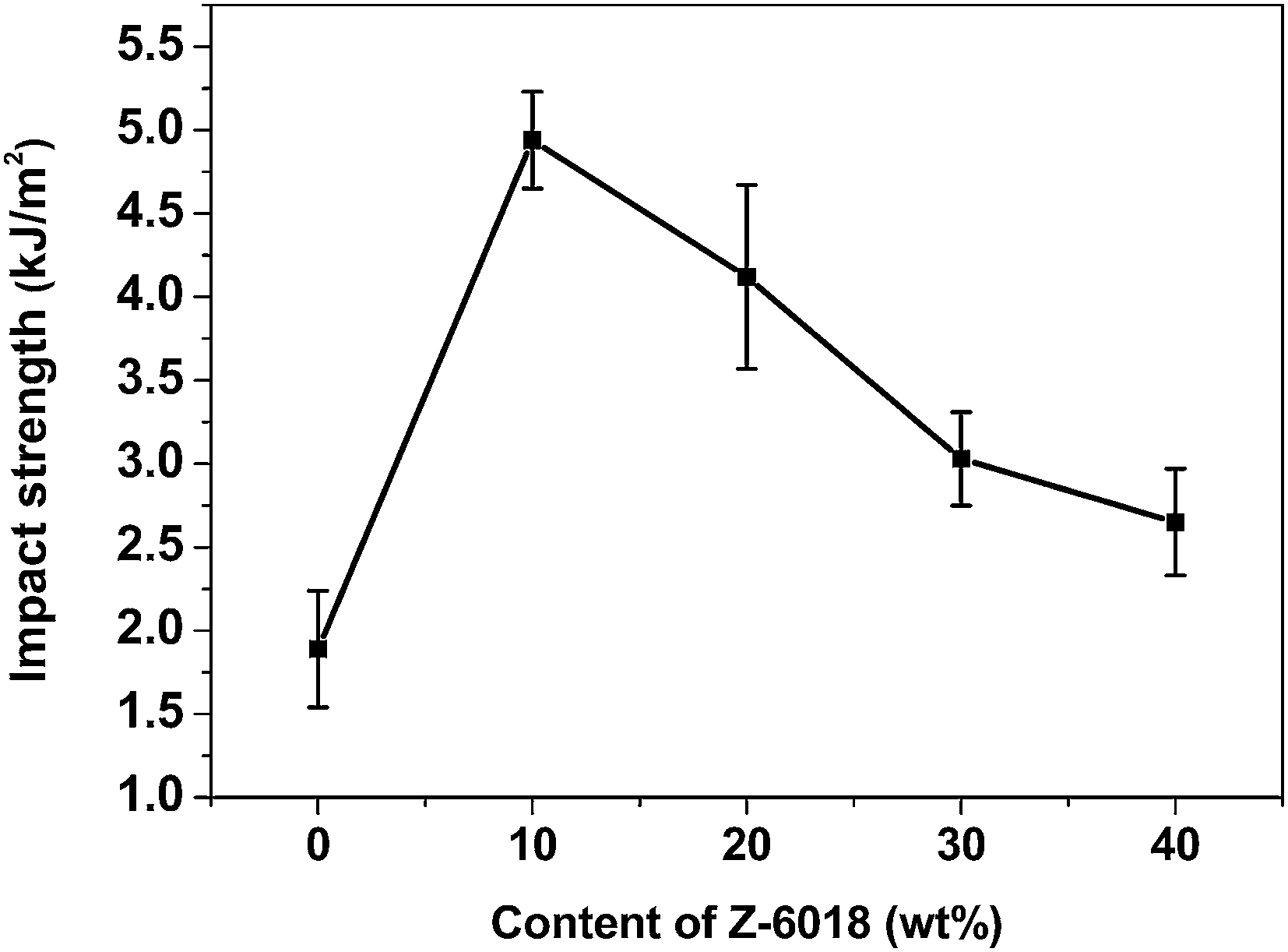 | ||
| Fig. 7 Impact strength of the PSDG systems as a function of Z-6018 content. | ||
It has been reported that the fracture toughness of a brittle epoxy is related to the size of the dispersion phase.36 The impact strength will initially increase with the increasing size of the dispersion phase to a maximum value and after that, fall with the further increasing of the second phase. Comparisons made between the SEM micro-graphs of the PSHG and PSDG systems reveal that, in the same dispersed phase content, the dispersed particle sizes for the PSDG systems are larger than those observed for the PSHG systems. Thus, the impact strength values of the PSHG systems are significantly increased with increasing SH023 content, while addition of Z-6018 increased first and then decreased.
3.4. TGA-FTIR analysis of cured epoxy mixture
In this work, TGA-FTIR was used to analyze the gas product during the thermal degradation process. The TGA and DTA curves of the PSHG and PSDG systems in nitrogen are presented in Fig. 8(a) and (b) and 9(a) and (b), respectively. The initial decomposition temperatures of the samples, which were evaluated by the temperature at 5 wt% weight loss (T−5%)37 and the solid residue left at 600 °C, were obtained from the TGA curve. The temperature at the maximum weight loss rate (Tmax1 and Tmax2) of the researched samples was obtained from the DTA curve. These data are given in Table 4.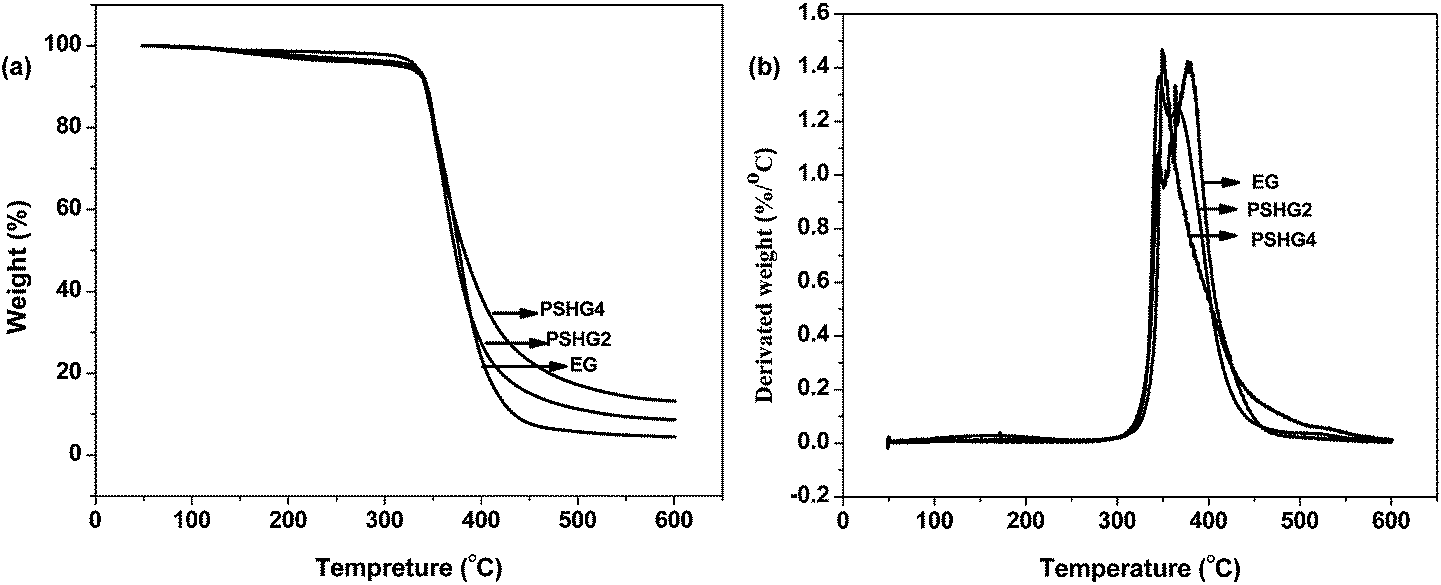 | ||
| Fig. 8 TGA(a) and DTA (b) cures of the PSHG systems under nitrogen at the indicated heating rates. | ||
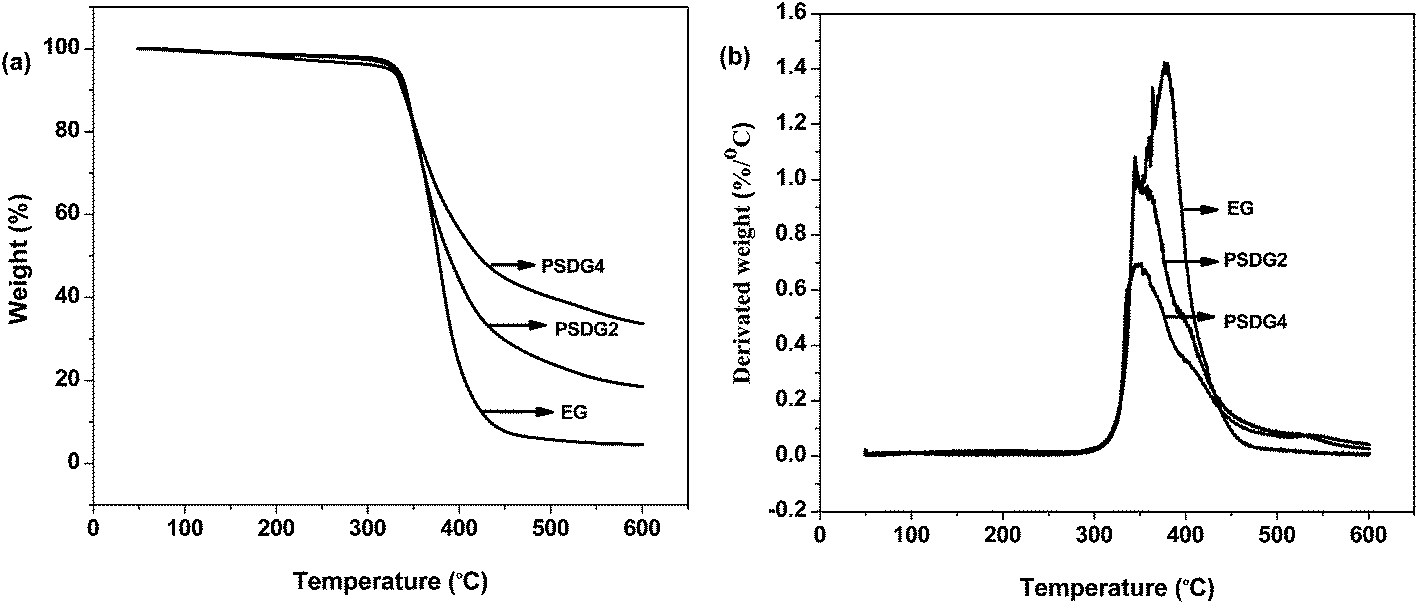 | ||
| Fig. 9 TGA (a) and DTA (b) curves of PSDG systems under nitrogen at the indicated heating rates. | ||
| Sample code | T−5% (°C) | Tmax1 (°C) | Tmax2 (°C) | Solid residue at 600 °C (%) |
|---|---|---|---|---|
| EG | 332.07 | 347.04 | 377.02 | 4.47 |
| PSHG2 | 329.31 | 346.85 | 361.99 | 8.64 |
| PSHG4 | 316.87 | — | 351.33 | 13.17 |
| PSDG2 | 328.88 | — | 344.89 | 18.44 |
| PSDG4 | 323.16 | — | 351.29 | 33.54 |
However, the thermal stability decreased according to the temperature of thermal degradation. Specifically, when the proportion of epoxy resin decreased, the systems began to lose weight earlier than the unmodified epoxy resins due to the lower cross-linking density of the cured systems. On the other hand, the siloxane bond –Si–O– has a binding energy of 445 kJ mol−1, which is higher than that of the carbon–carbon bond –C–C– in the epoxy resins. Consequently, a higher activation energy is required to destroy the siloxane inorganic polymer skeleton, thus leaving a higher solid residue.
As shown in Fig. 9(a), the solid residual yield increased to 33.54%, compared with epoxy resins with 40 wt% Z-6018. From the DTA curves of the PSDG systems in Fig. 9(b), it can be detected that the thermal decomposition temperature showed a similar trend for the PSDG and PSHG systems. In addition, a reduction of mass loss rate was detected with the increase of Z-6018 loading. These results could be explained by the stability of the inorganic nature of the siloxane structure and its partial ionic nature, which stabilized the epoxy resin from the heat. While heating, the low surface energy of silicone renders it to migrate to the surface of the epoxy resin by forming a protective self-heating layer with resistance to heat and the ability to slow down the thermal degradation of the polymer. Eventually, we find that epoxy resins modified by Z-6018 show a higher residual yield than epoxy resins modified by the same addition of SH023. This is due to the proportion of silicone in Z-6018 being higher than in SH023.
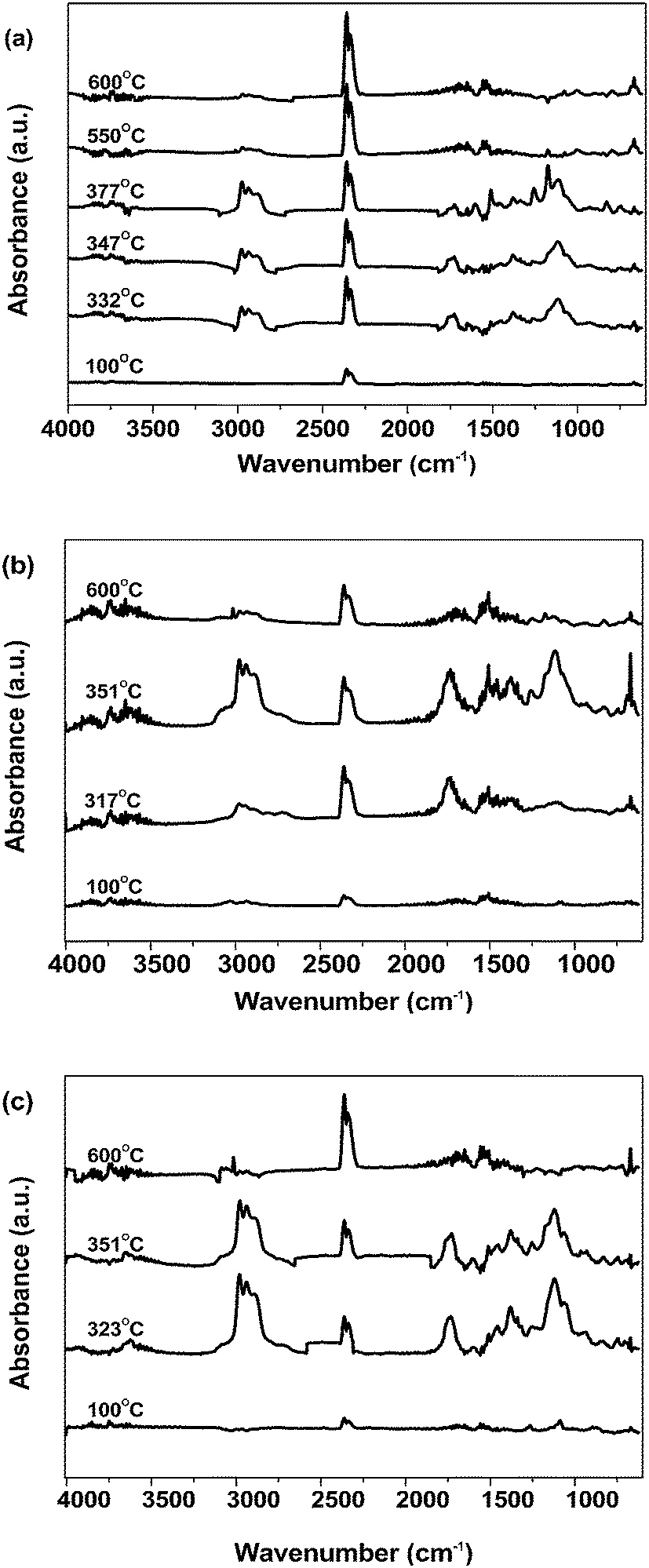 | ||
| Fig. 10 FTIR spectra of volatile products recorded at the indicated temperatures for the decomposition of silicone modified epoxy resins in a nitrogen atmosphere: (a) EG, (b) PSHG4, (c) PSDG4. | ||
During the second step of thermal degradation, the characteristic peak of water still existed until 55 min (550 °C). The resultant water could be associated with the dehydration of the two hydroxy propyl groups of the phenoxy resin. The evolved gases also show bands at 2990–3020 cm−1 and 1000–1300 cm−1 which are related to the sp2 and sp3 C–H stretching vibrations of para-disubstituted aromatic and aliphatic groups. The spectrum also revealed the presence of aldehyde-containing compounds by overlapping (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–H stretches (around 2700 cm−1 to 2900 cm−1 and 1724 cm−1) in the IR spectrum. The peaks that appeared at 3010 cm−1 and 1316 cm−1 are the characteristic peaks of CH4. The spectrum showed the appearance of bands at 1608 cm−1 at 340 °C and at 820 cm−1, which could be attributed to the CC stretching vibrations and the aromatic C–H stretching of para-disubstituted aromatic compounds, respectively. At 55 min (600 °C) the characteristic peaks of CO2 and N–H appeared and the thermal degradation rates of these products were very low.
O)–H stretches (around 2700 cm−1 to 2900 cm−1 and 1724 cm−1) in the IR spectrum. The peaks that appeared at 3010 cm−1 and 1316 cm−1 are the characteristic peaks of CH4. The spectrum showed the appearance of bands at 1608 cm−1 at 340 °C and at 820 cm−1, which could be attributed to the CC stretching vibrations and the aromatic C–H stretching of para-disubstituted aromatic compounds, respectively. At 55 min (600 °C) the characteristic peaks of CO2 and N–H appeared and the thermal degradation rates of these products were very low.
As shown in Fig. 10(b) and (c), the evolved gas analysis for PSHG4 or PSDG4 exhibited characteristic bands of compounds containing an –OH group (e.g. H2O, phenol; 3500–3900 cm−1), methyl substituted compounds (CH2/CH3 stretching, 2890–3000 cm−1), CO2 (2330, 2360 cm−1), compounds containing an aromatic ring (1320–1580 cm−1, 823 cm−1), hydrocarbons (C–H stretching at 1113 cm−1, 1248 cm−1 and 1172 cm−1), and N–C (675 cm−1).
From the above results, it is noted that the main evolved gas products for EG were different from epoxy resins modified by SH023 or Z-6018. The main evolved gas products of EG were composed of small molecules, such as CO2. In another aspect, epoxy resins modified by SH023 or Z-6018 released mainly methyl-substituted, carbonyl and aromatic gas products, which have higher molecular weights compared with that of the EG gas products.
4. Conclusion
In this paper, silicone-bridged epoxy resins were synthesized by reacting Z-6018 with epoxy resins. Epoxy resins blended with two different silicone resins (Z-6018, SH023) were investigated to compare their thermal and mechanical properties. The diameter of the silicone phases considerably increased with the addition of Z-6018 and SH023. The toughness of the silicone modified system increased considerably with a suitable diameter of silicone phases. The results showed that SH023 produced better enhancement of elongation at break and impact strength of epoxy resins. However, during thermal degradation, Z-6018 showed better residual yield contribution to epoxy resins than SH023 modified epoxy resins. The neat epoxy resins released compounds containing small molecules (CO2), methyl-substituted, carbonyl and aromatic ring compounds. The presence of Z-6018 or SH023 could retard the movement and scission of the main chain of EG during the thermal degradation process, thus resulting in a high residual yield.Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (51273118) and the Science & Technology Pillar Program of Sichuan (2013FZ0006) for financial support, and the Analytical and Testing Center of Sichuan University for providing SEM measurements.References
- D. Manjula, S. N. Jaisankar and P. Madhvesh, Eur. Polym. J., 2013, 49, 3561 CrossRef PubMed.
- R. Thomas, Y. Ding, Y. He, L. Yang, P. Moldenaers and W. Yang, Polymer, 2008, 49, 278 CrossRef PubMed.
- K. C. Jajam, M. M. Rahman and M. V. Hosur, Composites, Part A, 2014, 59, 57 CrossRef CAS PubMed.
- S. Khare, F. Khabaz and R. Khare, ACS Appl. Mater. Interfaces, 2014, 6, 6098 Search PubMed.
- M. Yu, J. Polym. Mater.: Sci. Eng., 2009, 150, 153 Search PubMed.
- L. H. Sinh, B. T. Son, N. N. Trung, D. G. Lim, S. Shin and J. Y. Bae, React. Funct. Polym., 2012, 72, 542 CrossRef CAS PubMed.
- H. El and A. Medhat, Metall. Mater. Trans. A, 2014, 45, 4046 CrossRef PubMed.
- S. Oprea, S. Vlad, A. Stanciu and M. Macoveanu, Eur. Polym. J., 2000, 36, 373 CrossRef CAS.
- D. K. Chattopadhyay, S. S. Panda and K. V. S. N. Raju, Prog. Org. Coat., 2005, 54, 10 CrossRef CAS PubMed.
- S. Chaudhary, S. Parthasarathy and D. Kumar, J. Appl. Polym. Sci., 2003, 87, 1562 CrossRef.
- K. P. O. Mahesh and M. ALagar, J. Appl. Polym. Sci., 2014, 131, 40490 Search PubMed.
- G. Tripathi and D. Srivastava, Mater. Sci. Eng., A, 2007, 443, 262 CrossRef PubMed.
- G. Tripathi and D. Srivastava, Mater. Sci. Eng., A, 2008, 496, 483 CrossRef PubMed.
- S. B. Chen, Q. H. Wang and T. M. Wang, Polym. Test., 2011, 7, 726 CrossRef PubMed.
- X. T. Yang, F. P. Yi, Z. R. Xin and S. X. Zheng, Polymer, 2009, 16, 4089 CrossRef PubMed.
- D. He, X. D. Ding, P. S. Chang and Q. M. Chen, Int. J. Adhes. Adhes., 2012, 38, 11 CrossRef CAS PubMed.
- N. Chikhi, S. Fellahi and M. Bakar, Eur. Polym. J., 2002, 38, 251 CrossRef CAS.
- K. S. Anandar, Z. Denchev and M. Alagar, Eur. Polym. J., 2006, 42, 2419 CrossRef PubMed.
- C. Li, C. M. Zuo, H. Fan, M. X. Yu and B. G. Li, Thermochim. Acta, 2012, 545, 75 CrossRef CAS PubMed.
- P. Murias, H. Maciejewski and G. Henryk, Eur. Polym. J., 2012, 48, 769 CrossRef CAS PubMed.
- W. J. Wang, L. H. Perng, G. H. Hsiue and F. C. Chang, Polymer, 2000, 41, 6113 CrossRef CAS.
- M. Ochi, K. Takemiya, O. Kiyolara and T. Nakanishi, Polymer, 1998, 39, 725 CrossRef CAS.
- M. Ochi, K. Takemiya, O. Kiyohara and T. Nakanishi, Polymer, 2000, 41, 195 CrossRef CAS.
- P. G. Liu, J. X. Song, L. H. He, X. Q. Liang, H. Y. Ding and Q. F. Li, Eur. Polym. J., 2008, 44, 940 CrossRef CAS PubMed.
- M. Ochi and S. Shimaoka, Polymer, 1999, 40, 1305 CrossRef CAS.
- M. Minoru, T. Akiyoshi, H. Katsumi and W. Akira, Composites, 1995, 26, 371 CrossRef.
- W. Liu, J. Kong, W. Eric Toh, R. Zhou, G. Ding, S. Huang, Y. Dong and X. Lu, Compos. Sci. Technol., 2013, 85, 1 CrossRef CAS PubMed.
- T. Y. Lo and S. K. Huang, J. Appl. Polym. Sci., 1998, 69, 1523 CrossRef CAS.
- A. G. Loera, F. Cara, M. Dumon and J. P. Pascault, Macromolecules, 2002, 35, 6291 CrossRef CAS.
- J. L. Hedrick, B. Haidar, T. P. Russell and D. C. Hofer, Macromolecules, 1998, 22, 1967 Search PubMed.
- J. L. Hedrick, T. P. Russell, B. Haidar and A. C. Tang, Macromolecules, 1989, 22, 4470 CrossRef CAS.
- A. Sharif, A. P. Gupta, S. Eram, A. Manawwer and S. K. Pandey, Prog. Org. Coat., 2005, 54, 248 CrossRef PubMed.
- S. J. Park, F. L. Jin and J. R. Lee, Macromol. Chem. Phys., 2004, 205, 2048 CrossRef CAS.
- Y. J. Woo, L. Y. Jin and L. S. Bok, Cryogenics, 2014, 61, 63 CrossRef PubMed.
- L. Deng, M. Shen, J. Yu, K. Wu and C. Ha, Ind. Eng. Chem. Res., 2012, 51, 8178 CrossRef CAS.
- D. J. Bray, P. Dittanet and F. J. Guild, Polymer, 2013, 54, 7022 CrossRef CAS PubMed.
- T. L. Lu, G. Z. Liang, Y. L. Peng and T. Chen, J. Appl. Polym. Sci., 2007, 106, 411 CrossRef.
- U. Braun, B. Schartel, M. A. Fichera and C. Jager, Polym. Degrad. Stab., 2007, 92, 1528 CrossRef CAS PubMed.
- R. R. Baker, S. Coburn, C. Liu and J. Tetteh, J. Anal. Appl. Pyrolysis, 2005, 74, 171 CrossRef CAS PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, Boston, 2nd edn, 1990 Search PubMed.
- A. I. Balabanovich, A. Hornung, D. Merz and H. Seifert, Polym. Degrad. Stab., 2004, 85, 713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09230a |
| This journal is © The Royal Society of Chemistry 2014 |