
Rheological and fluorescent properties of riboflavin–poly(N-isopropylacrylamide) hybrid hydrogel with a potentiality of forming Ag nanoparticle†
Abstract
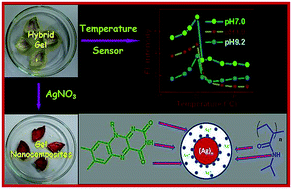
The riboflavin (R) and poly(N-isopropylacrylamide) (PNIPAAM) hybrid hydrogels have been prepared using the free radical polymerization of N-isopropylacrylamide in the presence of N,N′-methylene bisacrylamide as a cross linker for 1, 2 and 3 mM concentrations of R. The invariance of storage (G′) and loss (G′′) moduli over a wide range of angular frequency, where G′ > G′′ for R–PNIPAAM system characterize it to be behaving as a gel in the hybrid state. Both G′ and G′′ decrease with the increase of R concentration, but the decrease is four times higher in the former than in the latter. R–PNIPAAM gel has a higher critical strain value than PNIPAAM gels and it gradually increases with the increase in R concentration indicating that R is acting as a supramolecular cross-linker. The fluorescent intensity of R–PNIPAAM gels increases with the increase in R concentration and its variation with temperature at different pH shows an increase in the intensity value with temperature, showing the maximum at ∼30 °C because of the coil-to-globule transition of PNIPAAM chains, suggesting that the R–PNIPAAM gel can be used to be a probe for temperature detection. The sensitivity index of the fluorescent temperature sensing of R–PNIPAAM gel is moderate and it is highest at pH 7. R becomes gradually released from the R–PNIPAAM gel when dipped into water (pH 7) at 20 °C, initially at a slower rate, then at a higher rate, and finally it shows a leveling for the release of 19% of embedded R at 24 h of aging. Silver nanoparticles that are grown in the R–PNIPAAM gels by immersing the slashed gel pieces in AgNO3 solution stay at both the surface and the interior of the R–PNIPAAM fibres, maintaining the gel structure intact with a decrease in critical strain and causing a quenching of the fluorescence intensity of R–PNIPAAM gels. The average size and size distribution of AgNPs linearly increase with R concentration at a constant AgNO3 concentration.
 Please wait while we load your content...
Please wait while we load your content...

