
Syntheses of indolizinones from an intramolecular one-pot process of gem-dibromoolefins†
Fei Tanga,
Chaonan Chen*a,
Yiqian Zhoua,
Cai Lina and
Jiancun Zhang*ab
aGuangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, 190 Kai Yuan Avenue, Science Park, Guangzhou 510530, PR China. E-mail: chen_chaonan@gibh.ac.cn; zhang_jiancun@gibh.ac.cn
bState Key Laboratory of Respiratory Disease, Guangzhou Medical College, Guangzhou, PR China
First published on 30th September 2014
Abstract
Transition-metal-catalyzed C–H activation has recently emerged as a powerful tool for the syntheses of natural products and bioactive compounds. We developed an efficient sequential one-pot intramolecular C–N bond formation and direct C–H arylation method to construct a series of unusual indolizinone scaffolds using gem-dibromoolefins in moderate to good yields under mild conditions.
Transition-metal-catalyzed C–H activation has emerged as a powerful tool for the formation of carbon–carbon and carbon–heteroatom bonds.1 A C–H activation strategy has been successfully applied for the syntheses of natural products and bioactive compounds.2 With selective C–H activation in the aromatic and heteroaromatic compounds, some complex polycyclic heterocycles have been constructed efficiently.3 One-pot C–H activation coupling with other reactions would be more efficient and useful for the construction of many structurally interesting motifs.4
gem-Dibromoolefins, as bidentate electrophiles, have been found to be versatile intermediates in constructing various heterocyclic motifs.5 With catalyzed inter- or intramolecular nucleophilic reactions, such as Heck,6 Ullmann,7 Sonogashira,8 Suzuki9 and Buchwald10 reactions, two new C–C and/or C–N bonds can be formed in one step by using gem-dibromoolefins as the building block. Many facile methods for the formation of polycyclic heteroaromatics have been developed from gem-dibromoolefins.5a,d–f,6–10 In conjunction with our study of gem-dibromoolefins for the syntheses of heterocycles,11 we have developed a facile and efficient method to construct indolizinone compounds via an intramolecular one-pot amination and C–H activation process with gem-dibromoolefins through an unexpected cyclization mode. Most of the compounds obtained are novel polycyclic heterocycles, and may be of great interest in drug discovery.
We initiated our investigation aiming for construction of the camptothecin framework by employing pyridinyl compound (1a) as the precursor for the synthesis of indolizinone 6. As the C–H functionalization of pyridines moiety is often hampered by its poor electron density of the aromatic ring,12 we used the pyridine N-oxides in order to activate the o-H of pyridines to facilitate the subsequent C–H insertion step (Scheme 1).13 We first studied the reaction conditions with substrate 1b to investigate the effects of different catalysts, ligands, bases, solvents and temperatures for the preparation of 5 and subsequently 6 upon reduction of the N-oxides. The results are shown in Table 1.
 | ||
| Scheme 1 Planned for the synthesis of pyridine-fused indolizinone derivatives. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yieldc [%] |
| a Reaction were carried out under an argon atmosphere.b Catalyst (10 mol%), ligand (20 mol%), 120 °C.c Yields of compound 2b, determined by NMR adding mesitylene as the internal standard.d Yield of isolated product.e Without catalyst and ligand and react in the air. | |||||
| 1b | Pd(OAc)2 | PtBu3–HBF4 | K2CO3 | Toluene | Trace |
| 2b | Pd(OAc)2 | PCy3 | Cs2CO3 | Toluene | Trace |
| 3b | Pd2(dba)3 | P(2-MeOPh)3 | Cs2CO3 | Toluene | Trace |
| 4 | CuI | Phen | Cs2CO3 | Toluene | 54 |
| 5 | CuI | Phen | K3PO4 | Toluene | 73 |
| 6 | CuI | Phen | K2CO3 | Toluene | 77d |
| 7 | CuI | L-Proline | K2CO3 | Toluene | 6.5 |
| 8 | CuCl | Phen | K2CO3 | Toluene | 65 |
| 9 | CuBr | Phen | K2CO3 | Toluene | 45 |
| 10 | CuI | Phen | K2CO3 | THF | 42 |
| 11 | CuI | Phen | K2CO3 | 1,4-Dioxane | 64 |
| 12 | CuI | Phen | K2CO3 | CH3CN | 17 |
| 13e | K2CO3 | Toluene | 99d | ||

After an extensive survey of reaction conditions, we couldn't obtain the desired product by using various Pd or Cu catalysts. Quite surprisingly, compound 2b rather than the expected compound 2c was formed in moderate yield (Table 1, entry 6) and the structure of this intermediate was confirmed by two-dimensional NMR NOE spectroscopy experiment. Later, we found that the yield of 2b increased significantly when only base was added to the reaction (entry 13). Although the desired indolizinone compound 5 couldn't be afforded in this condition, a trace amount of another structurally rare type of indolizinone structure 3b was obtained instead when Pd catalysts were present. We then started to investigate the reaction conditions of transforming 2b into the indolizinone compound 3b separately and the results are summarized in Table 2. After we screened several reaction parameters, the reaction system with Pd(OAc)2 (10 mol%), P(2-MeOPh)3 (20 mol%), and K2CO3 (3.0 equiv.) in toluene was found to be most efficient (Table 2, entry 5). Finally we combined these two steps into a one-pot process. After the intramolecular amination addition reaction completed with mild base under 70 °C, we added catalyst and ligand into the reaction mixture for the direct arylation while raising the temperature to 120 °C for the C–H insertion reaction. The desired compound 3b was afforded in good yield through this sequential one-pot process. 3b was hydrogenated by Raney Ni to afford compound 3a in excellent yield (Scheme 2).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Yieldb [%] |
| a Reactions were carried out under an argon atmosphere.b Yields were determined by NMR adding mesitylene as the internal standard.c Yield of isolated product.d 1 equiv. of Bu4NBr was added. | ||||
| 1 | Pd(OAc)2 | PCy3 | K2CO3 | 54 |
| 2 | Pd(OAc)2 | PCy3 | Cs2CO3 | 37 |
| 3 | Pd(OAc)2 | PCy3 | K3PO4 | 61 |
| 4 | Pd(OAc)2 | PtBu3·HBF4 | K2CO3 | 11 |
| 5 | Pd(OAc)2 | P(2-MeOPh)3 | K2CO3 | 91c |
| 6 | Pd(OAc)2 | Diethyl phosphite | K2CO3 | 8 |
| 7 | Pd(OAc)2 | P(2-MeOPh)3 | K2CO3 | 0d |
| 8 | Pd(OAc)2 | PPh3 | K2CO3 | 79 |
| 9 | Pd(OAc)2 | JohnPhos | K2CO3 | 39 |
| 10 | Pd(OAc)2 | P(2-MeOPh)3 | K3PO4 | 74 |
| 11 | Pd2(dba)3 | P(2-MeOPh)3 | K2CO3 | 40 |
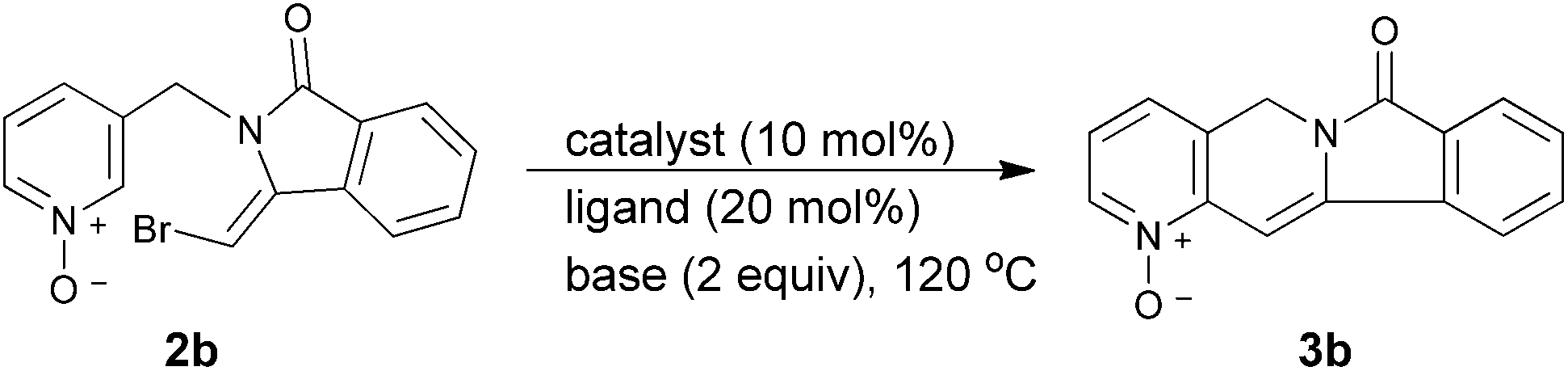
 | ||
| Scheme 2 Reduction of indolizinone N-oxide. | ||
With this optimized reaction conditions in hand, we set to investigate the generality of this one-pot reaction for various pyridinyl heterocycles (Table 3). In the case of unoxidized pyridinyl substrate, the reaction underwent smoothly but two isomers 3a and 4 from o and p insertions were obtained in 30% and 60% of yields respectively. We started by introducing substitutions on pyridine to study the substitution effects. Methyl substituted pyridinyl compounds were well tolerated (3c, 3d). We extended the linker between the pyridine and the nitrogen atom, and the pyridine-fused six-membered ring was formed in 63% under this standard condition (3e). A limitation of this protocol was observed for substrate 1f, as the m-H of the pyridine couldn't be activated under the standard conditions with only the intermediate 2f obtained.
|
|---|
| a Yield of isolated product. |
 |

We then expanded the method to other aromatics. Benzene and substituted benzenes were chosen as the substrates first. We found that the reaction was sluggish in the amination addition step and complete conversion could not be reached even after several days. We reasoned that it may due to the lack of basicity of K2CO3. We then added 1 eq. of KOtBu to the reaction mixture, the amination addition step was finished in about one hour and the desired products were afforded readily. Benzene with various of functional group substitutions were well tolerated including both electron-donating and electron-withdrawing groups. Besides benzene aromatics, other aromatic rings including furan, thiophene, indol, quinolone, naphthalene moieties were applied to the reaction and the desired product were obtained in moderate to good yields. The results were summarized in Table 4.
|
|---|
| a Yield of isolated product. |
 |

A tentative mechanism of the reaction is depicted in Scheme 3. The first step involves a simple elimination reaction to form the acetylene bromide followed by a 5-exo intramolecular N-nucleophilic addition as previously reported.14 The monobromo olefinic compound intermediate 2b was isolated and its structure was confirmed. This monobromo intermediate undergoes C–Br bond insertion with Pd followed by intramolecular C–H activation and insertion to give the indolidizines.15
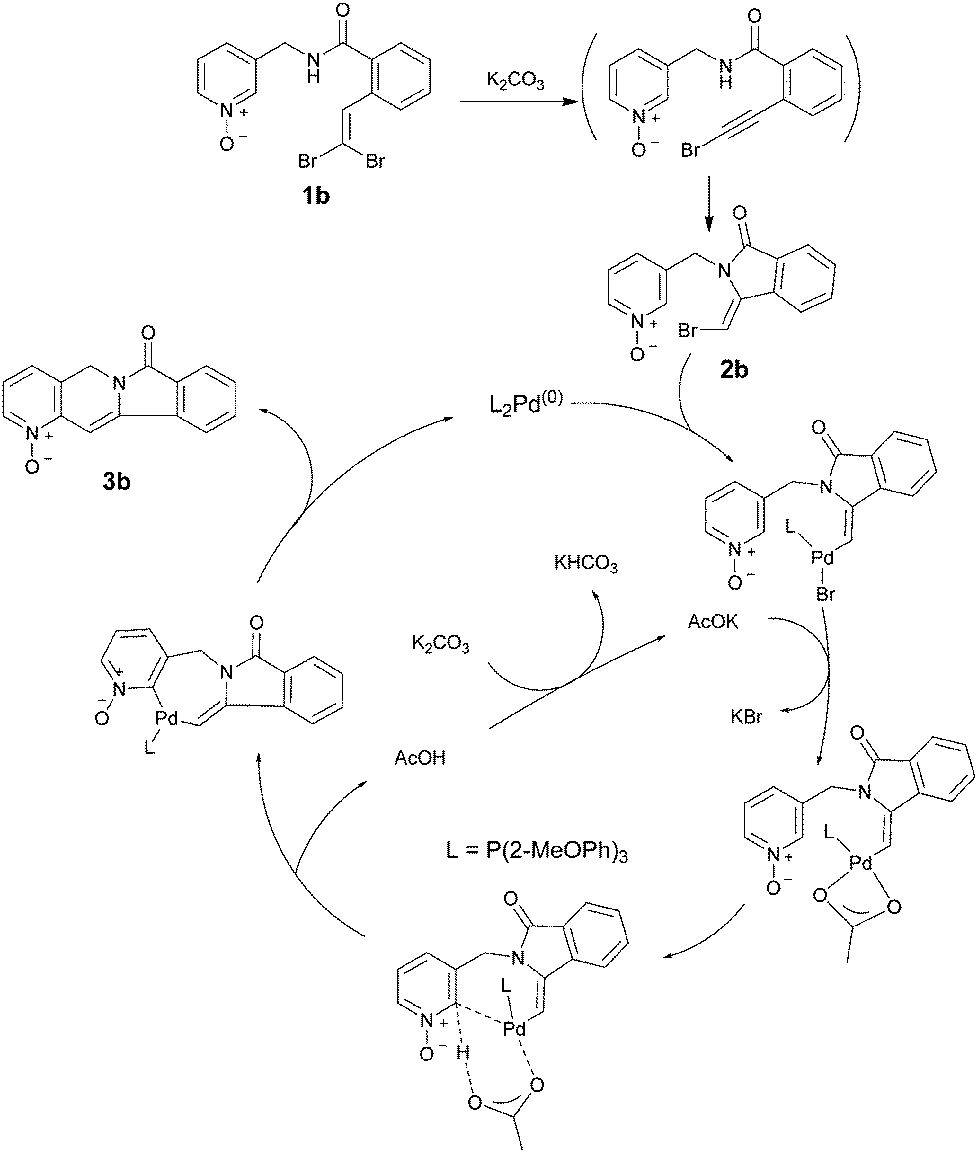 | ||
| Scheme 3 A proposed mechanism for this sequential addition and cross-coupling process. | ||
In summary, we have developed an efficient and practical intramolecular N-nucleophilic addition and direct C–H arylation process using gem-dibromoolefins for the synthesis of uncommon indolizinone scaffold. This reaction process is applicable to various aromatic rings with various substitutions, and a series of novel indolizinone compounds were obtained in good yields. The method provides an entry into the efficient preparations of uncommon indolizinone structures.
Typical experimental process
3-((2-(2,2-Dibromovinyl)benzamido)methyl)pyridine N1-oxide (1b, 100 mg, 0.24 mmol, 1.0 equiv.) was dissolved in toluene and K2CO3 (100 mg, 0.74 mmol, 3.0 equiv.) was added. The mixture was stirred at 70 °C for 5 h. Then Pd(OAc)2 (5.4 mg, 0.024 mmol, 10 mol%) and P(2-MeOPh)3 (17 mg, 0.048 mmol, 20 mol%) were added. The reaction mixture was stirred under an argon atmosphere at 120 °C for 12 h. The resulting mixture was concentrated in vacuo and purified by column chromatography on silica gel to give the product 3b (55 mg, 90%).Acknowledgements
The research was supported in part by Foshan municipal funds for scientific and technological innovation projects (2013HK100012), Guangzhou Science and Technology Project (2014J4100222), Chinese NSFC grant (nos 21402205).Notes and references
- (a) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed; (b) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2010, 111, 1293 CrossRef PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed.
- (a) W. Ma, K. Graczyk and L. Ackermann, Org. Lett., 2012, 14, 6318 CrossRef CAS PubMed; (b) L.-C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253 RSC; (c) M. A. Campo, Q. Huang, T. Yao, Q. Tian and R. C. Larock, J. Am. Chem. Soc., 2003, 125, 11506 CrossRef CAS PubMed; (d) J. Huang, C. Wan, M.-F. Xu and Q. Zhu, Eur. J. Org. Chem., 2013, 1876 CrossRef CAS; (e) Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306 CrossRef CAS PubMed; (f) R. Xia, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2012, 14, 5546 CrossRef CAS PubMed.
- (a) H. A. Wegner, L. T. Scott and A. de Meijere, J. Org. Chem., 2003, 68, 883 CrossRef CAS PubMed; (b) R. B. Bedford and M. Betham, J. Org. Chem., 2006, 71, 9403 CrossRef CAS PubMed; (c) J.-P. Leclerc, M. André and K. Fagnou, J. Org. Chem., 2006, 71, 1711 CrossRef CAS PubMed; (d) A. Martins, D. Alberico and M. Lautens, Org. Lett., 2006, 8, 4827 CrossRef CAS PubMed; (e) L. F. Tietze and F. Lotz, Eur. J. Org. Chem., 2006, 4676 CrossRef CAS; (f) L. Ackermann and A. Althammer, Angew. Chem., Int. Ed., 2007, 46, 1627 CrossRef CAS PubMed; (g) T. Watanabe, S. Ueda, S. Inuki, S. Oishi, N. Fujii and H. Ohno, Chem. Commun., 2007, 4516 RSC; (h) T. Jensen, H. Pedersen, B. Bang-Andersen, R. Madsen and M. Jørgensen, Angew. Chem., Int. Ed., 2008, 47, 888 CrossRef CAS PubMed; (i) A. K. Yadav, S. Verbeeck, S. Hostyn, P. Franck, S. Sergeyev and B. U. W. Maes, Org. Lett., 2013, 15, 1060 CrossRef CAS PubMed; (j) K. Beydoun and H. Doucet, Eur. J. Org. Chem., 2012, 6745 CrossRef CAS.
- (a) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2010, 132, 11416 CrossRef CAS PubMed; (b) Z.-J. Wang, J.-G. Yang, F. Yang and W. Bao, Org. Lett., 2010, 12, 3034 CrossRef CAS PubMed; (c) Z.-J. Wang, F. Yang, X. Lv and W. Bao, J. Org. Chem., 2011, 76, 967 CrossRef CAS PubMed; (d) G. Chelucci, Chem. Rev., 2012, 112, 1344 CrossRef CAS PubMed; (e) R. Y. Huang, P. T. Franke, N. Nicolaus and M. Lautens, Tetrahedron, 2013, 69, 4395 CrossRef CAS PubMed; (f) M. L. N. Rao, D. N. Jadhav and P. Dasgupta, Eur. J. Org. Chem., 2013, 781 CrossRef CAS.
- C. S. Bryan and M. Lautens, Org. Lett., 2010, 12, 2754 CrossRef CAS PubMed.
- H. Xu, Y. Zhang, J. Huang and W. Chen, Org. Lett., 2010, 12, 3704 CrossRef CAS PubMed.
- M. Nagamochi, Y.-Q. Fang and M. Lautens, Org. Lett., 2007, 9, 2955 CrossRef CAS PubMed.
- N. Nicolaus, P. T. Franke and M. Lautens, Org. Lett., 2011, 13, 4236 CrossRef CAS PubMed.
- C. S. Bryan and M. Lautens, Org. Lett., 2008, 10, 4633 CrossRef CAS PubMed.
- (a) B. Jiang, K. Tao, W. Shen and J. Zhang, Tetrahedron Lett., 2010, 51, 6342 CrossRef CAS PubMed; (b) K. Tao, J. Zheng, Z. Liu, W. Shen and J. Zhang, Tetrahedron Lett., 2010, 51, 3246 CrossRef CAS PubMed; (c) P. Liu, K. Tao, L. Zhao, W. Shen and J. Zhang, Tetrahedron Lett., 2012, 53, 560 CrossRef CAS PubMed.
- M. Wasa, B. T. Worrell and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 1275 CrossRef CAS PubMed.
- (a) A. T. Londregan, S. Jennings and L. Wei, Org. Lett., 2011, 13, 1840 CrossRef CAS PubMed; (b) S. Zhang, L.-Y. Liao, F. Zhang and X.-F. Duan, J. Org. Chem., 2013, 78, 2720 CrossRef CAS PubMed; (c) J. T. Myers and J. M. Hanna Jr, Tetrahedron Lett., 2012, 53, 612 CrossRef CAS PubMed; (d) A. T. Londregan, K. A. Farley, C. Limberakis, P. B. Mullins and D. W. Piotrowski, Org. Lett., 2012, 14, 2890 CrossRef CAS PubMed; (e) W. J. Lominac, M. L. D'Angelo, M. D. Smith, D. A. Ollison and J. M. Hanna Jr, Tetrahedron Lett., 2012, 53, 906 CrossRef CAS PubMed; (f) X. Gong, G. Song, H. Zhang and X. Li, Org. Lett., 2011, 13, 1766 CrossRef CAS PubMed; (g) S. Duric and C. C. Tzschucke, Org. Lett., 2011, 13, 2310 CrossRef CAS PubMed; (h) A. T. Londregan, S. Jennings and L. Wei, Org. Lett., 2010, 12, 5254 CrossRef CAS PubMed; (i) J. M. Keith, J. Org. Chem., 2010, 75, 2722 CrossRef CAS PubMed; (j) O. V. Larionov, D. Stephens, A. Mfuh and G. Chavez, Org. Lett., 2014, 16, 864 CrossRef CAS PubMed.
- (a) A. Coste, G. Karthikeyan, F. Couty and G. Evano, Angew. Chem., Int. Ed., 2009, 48, 4381 CrossRef CAS PubMed; (b) C. Wang, C. Sun, F. Weng, M. Gao, B. Liu and B. Xu, Tetrahedron Lett., 2011, 52, 2984 CrossRef CAS PubMed; (c) J. Liu, F. Dai, Z. Yang, S. Wang, K. Xie, A. Wang, X. Chen and Z. Tan, Tetrahedron Lett., 2012, 53, 5678 CrossRef CAS PubMed; (d) Z. Huang, R. Shang, Z.-R. Zhang, X.-D. Tan, X. Xiao and Y. Fu, J. Org. Chem., 2013, 78, 4551 CrossRef CAS PubMed; (e) C. J. Ball and M. C. Willis, Eur. J. Org. Chem., 2013, 425 CrossRef CAS; (f) Y. Zhu, P. Sun, H. L. Yang, L. H. Lu, H. Yan, M. Creus and J. C. Mao, Eur. J. Org. Chem., 2012, 4831 CrossRef CAS; (g) S. E. Kiruthika and P. T. Perumal, Org. Lett., 2014, 16, 484 CrossRef CAS PubMed.
- H.-Y. Sun, S. I. Gorelsky, D. R. Stuart, L.-C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, spectral data. See DOI: 10.1039/c4ra09206f |
| This journal is © The Royal Society of Chemistry 2014 |