
Acceptorless dehydrogenative synthesis of 2-substituted quinazolines from 2-aminobenzylamine with primary alcohols or aldehydes by heterogeneous Pt catalysts†
Abstract
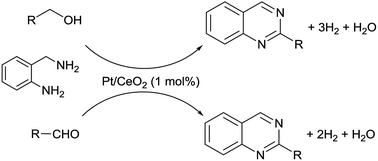
CeO2-supported Pt nanoparticle catalysts enabled the acceptorless dehydrogenative synthesis of 2-substituted quinazolines from 2-aminobenzylamine with aliphatic and benzyl primary alcohols or aldehydes with low catalyst loading, wide substrate scope and good catalyst reusability, demonstrating the first acceptor-free and additive-free catalytic system for this reaction.
 Please wait while we load your content...
Please wait while we load your content...

