
Structural evolution, sequential oxidation and chemical bonding in tri-yttrium oxide clusters: Y3Ox− and Y3Ox (x = 0–6)†
Lei Xua,
Chan-Juan Xiaa,
Ling-Fei Wanga,
Lu Xiea,
Bin Wanga,
Yong-Fan Zhang*ab and
Xin Huang*ab
aCollege of Chemistry, Fuzhou University, Fuzhou, Fujian 350116, PR China. E-mail: xhuang@fzu.edu.cn; zhangyf@fzu.edu.cn; Fax: +86 591 22866156; Tel: +86 591 22866139
bFujian Provincial Key Laboratory of Theoretical and Computational Chemistry, Xiamen, Fujian 361005, PR China. E-mail: xhuang@fzu.edu.cn; zhangyf@fzu.edu.cn
First published on 27th October 2014
Abstract
We report a systematic and comprehensive investigation on the electronic structures and chemical bonding of a series of tri-yttrium oxide clusters, Y3Ox−/0 (x = 0–6), using density functional theory (DFT) calculations. The generalized Koopmans' theorem was applied to predict the vertical detachment energies (VDEs) and simulate the photoelectron spectra (PES) for Y3Ox− (x = 0–6) clusters. A trend of sequential oxidation is observed from Y3O− to Y3O6−. All of these clusters tend to form structures with a capped oxygen atom. For Y3Ox−/0 (x = 2–4), the O atoms prefer the bridging sites of Y3O−/0, whereas the fifth O atoms for Y3O5−/0 are bonded to the terminal sites. In particular, the ground states of Y3O6−/0 are found to be interesting species, which may be viewed as molecular models for dioxygen activation by Y3O4−/0. σ- and π-aromaticity is found in Y3− by the molecular orbital analysis and Adaptive Natural Density Partitioning (AdNDP) analysis. Molecular orbital analyses are performed to analyze the chemical bonding in the tri-yttrium oxide clusters and to elucidate their electronic and structural evolution.
1. Introduction
Yttrium oxides have important applications in the field of catalysts, fireproofing, electrode materials, and magnetic materials,1–11 and they are especially used as doping agents for catalyst supports and catalysts.8–11 For instance, yttria-doped alumina is more thermally stable than bare alumina and can be used as a catalyst support for the thermo-catalytic cracking process.8,9 Furthermore, homonuclear yttrium oxide clusters and heteronuclear aluminum-doped yttrium oxide clusters have exhibited different catalytic performances in redox reactions with N2O/CO. The yttrium oxide cluster doped with aluminum (YAlO3+˙) was proven to bring about efficient CO oxidation, and the reaction pattern of YAlO3+˙ is considerably different from that of Y2O3+˙ as a result of the doping effects.10 As a first step in developing a comprehensive understanding of complex catalytic processes on early transition-metal oxides, we are interested in systematically studying the electronic structure and chemical bonding of isolated transition-metal oxide clusters.Yttrium oxide clusters have been studied both experimentally10,12–14 and theoretically.10,11,15–18 The photoionization spectra of yttrium clusters Yn and YnO (n = 2–31) have been recorded by Knickelbein, and a steep non-monotonic decrease of ionization potentials (IPs) was discovered up to about ten atoms, followed by a more gradual decline.12 Wu and Wang13 reported the photoelectron spectrum and electronic structures of YOn− (n = 1–5). Pramann and co-workers14 obtained the photoelectron spectrum of YnOm (n = 2–10, m = 1–3), in which a shifting of the threshold energies to higher binding energies was observed with the increase of cluster size n. Recently, attentions have also been focused on the theoretical studies of yttrium oxide clusters. For example, the structural and electronic properties of Y3O and Y4O were studied by Gu et al., where the structures of Y3O and Y4O were found to be pyramidal and bi-pyramidal, respectively, and the calculated electron affinities and ionization potentials were in good agreement with the available experimental data.15,16 In addition, Yang and co-workers investigated the structural, electronic, and magnetic properties of YnO (n = 2–14).17 The calculated ionization potentials and electron affinities were in good agreement with the experimental results, implying that the ground states of these clusters are reliable to better understand yttrium oxide clusters and materials. Very recently, Rahane et al.18 reported a DFT study on the structural and electronic properties of (Y2O3)n0/±1 (n = 1–10) clusters and found that the charge transfer from the Y atoms to the oxygen atoms increases with the increase in cluster size. To the best of our knowledge, there are still very few investigations on tri-yttrium oxide clusters.
We have been interested in developing cluster models to characterize metal cluster species in the gas phase and to provide fundamental mechanistic insights for catalysts,19–24 as well as novel all-metal aromaticity.25 In the previous studies, all-metal aromaticity was first reported using Al42− as an example with double σ- and π-aromaticity.26 New computational evidence has been presented about triple (σ-, π- and δ-) aromaticity in the lowest D3h (1A′1) singlet state of Hf3.27 Nowadays, a few review articles concerning all-transition-metal aromaticity/antiaromaticity have been published.28–35 For example Boldyrev et al.28 performed the adaptive natural density partitioning (AdNDP) method to state the aromaticity for the Sc3− (D3h 1A′1) cluster. σ- and π-aromaticity represents a mode of chemical bonding and may play an important role in multinuclear transition metal compounds.
In this work, we focus on exploring the geometric and electronic structures of early transition metal clusters and their oxide clusters. DFT calculations are employed to elucidate the electronic structure and chemical bonding of Y3Ox−/0 (x = 0–6). In particular, the ground states of Y3O6−/0 are found to be interesting species, which may be viewed as molecular models for dioxygen activation by Y3O4−/0. A sequential oxidation is observed in the Y3Ox− (x = 0–6) clusters as a function of x. The AdNDP method is performed to analyze the aromaticity of Y3− cluster, indicating that the Y3− possesses σ- and π-aromaticity.
2. Computational methods
The theoretical calculations were performed at the DFT level using the BP86 functional.36,37 The global minimum searches were performed using analytical gradients with the Stuttgart relativistic small core basis set and effective core potential38,39 augmented with two f-type and one g-type polarization function for yttrium [ζ(f) = 0.144, 0.546; ζ(g) = 0.249] as recommended by Martin and Sundermann40 and the aug-cc-pVTZ basis set41,42 for oxygen. Scalar relativistic effects, that is, the mass velocity and Darwin effects, were taken into account via the quasi-relativistic pseudo-potentials. Vibrational frequency calculations were performed to verify the nature of the stationary points. The vertical electron detachment energies (VDEs) were calculated using the generalized Koopmans' theorem by adding a correction term to the eigen values of the anion.43 The correction term was calculated as δE = E1 − E2 − εHOMO, where E1 and E2 are the total energies of the anion and neutral ion, respectively, in their ground states at the anion equilibrium geometry, and εHOMO corresponds to the eigen value of the highest occupied molecular orbital (HOMO) of the anion. The relative energies of the low-lying structures were further evaluated via single-point calculations at the coupled cluster [CCSD(T)]44–48 level with the Y/Stuttgart + 2f1g/O/aug-cc-pVTZ basis sets at the BP86 geometries. For open-shell systems, the UCCSD(T) approach was used, where a restricted open-shell Hartree–Fock (ROHF) calculation was initially performed and the spin constraint was relaxed in the correlation treatment. In addition, more accurate optimizations in CCSD(T) were carried out for certain close-lying structures with the Y/Stuttgart + 2f1g/O/aug-cc-pVTZ basis sets. All the DFT calculations were performed with the Gaussian 09 software package,49 and the CCSD(T) calculations were done using the MOLPRO 2010.1 package.50 Three-dimensional contours of the molecular orbitals were visualized using the VMD software.51 Different exchange–correlation functionals were tested for accuracy and consistency. The BP86 functional showed superior results in terms of electron binding energies and structures for yttrium oxide clusters. The previous studies on yttrium oxide clusters52,53 also indicated that BP86 calculations can give good consistency with the experimental results and better overall performance in predicting the molecular properties. Therefore, we used the results with the BP86 functional for our discussion.The adaptive natural density partitioning (AdNDP) method has been recently developed by Boldyrev et al.28 The principles of the method have been described elsewhere.54 The AdNDP analysis for the ground state of Y3− cluster was performed using DFT at the BP86/lanl2dz level. The results of the AdNDP analysis were visualized using the MOLEKEL 5.4 software.55
3. Theoretical results
The optimized ground states and low-lying isomers (within ∼0.40 eV) at the BP86 level for the anionic Y3Ox− and neutral Y3Ox (x = 0–6) are presented in Fig. 1–7, respectively. More optimized geometries at higher energies are given as ESI (Fig. S1–S7†). In the following discussion, Ot, Ob and Oc represent the terminal, bridging and capped oxygen atoms, respectively.3.1 Y3− and Y3
For the tri-yttrium clusters, four possible atomic arrangements: linear (C∞v or D∞h), equilateral triangle (D3h), isosceles triangle (C2v), or completely distorted triangle (Cs) were considered. The ground states and the selected low-lying isomers (within ∼0.40 eV) of the tri-yttrium clusters (Y3− and Y3) are listed in Fig. 1. Our theoretical results clearly show that the global minimum of Y3− is a D3h (1A′1) structure (Fig. 1a), which is in accordance with the previous study by Chi et al.31 The corresponding triplet (D3h, 3A′′2) (Fig. 1b) and (D3h, 3A′1) (Fig. 1c) are 0.27 eV and 0.34 eV higher in energy, respectively. The low-lying isomer (Fig. 1d) with a C2v (3A2) symmetry is 0.34 eV higher above the ground state. | ||
| Fig. 1 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
For the neutral Y3, the ground state is revealed to be D3h (2A′1) (Fig. 1e). The Y–Y bond is slightly longer (∼0.08 Å) compared to that of the anion (Fig. 1a). In the work by Dai et al.,56 the structure (D3h, 2A′′) (Fig. 1f) was predicted to be the ground state of Y3 using the complete active space multiconfiguration self-consistent field (CAS-MCSCF) method. However, it was 0.10 eV higher in energy above the D3h (2A′1) state in our calculation. The corresponding quartet (D3h, 4A′′2) (Fig. 1g) is 0.27 eV higher in energy at the BP86 level. Similar to the anion, there also exists a C2v (4B2) structure, which is 0.31 eV above the ground state. The single-point CCSD(T) (Table SI†) were also carried out, and the results are in agreement with that of our BP86 calculations instead of that of the previous study.56 Further theoretical calculations with more sophisticated methods may be necessary to resolve the true ground state of Y3.
3.2 Oxygen-deficient clusters: Y3Ox−/0 (x = 0–4)
 | ||
| Fig. 2 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3O−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
The neutral ground state (C3v, 2A1) (Fig. 2h) is totally conformed to the result of Yang et al.,17 which resembles that of Y3O−, except for a slightly lengthened Y–Y bond (by 0.061 Å) and shortened Y–O bond (by 0.016 Å). The low-lying isomer (Fig. 2i) with C2v symmetry is a quartet (4B1), and 0.25 eV above the ground state.
 | ||
| Fig. 3 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3O2−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
The global minimum (Cs, 2A′) (Fig. 3h) for the neutral Y3O2 is similar to that of Y3O2−. The capped oxygen atom is closer to the yttrium atoms at the bottom compared to Y3O2−. Three low-lying isomers are found within 0.20 eV at the BP86 level. Similar to the anion, single-point CCSD(T) calculations are performed for these isomers, showing that isomer (Cs, 2A′) (Fig. 3h) is the ground state. It is lower by at least 0.31 eV in energy than the others, as shown in Table SI.†
 | ||
| Fig. 4 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3O3−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
Similar to the anion, the ground state of neutral Y3O3 has a capped and two bridging O atoms, as shown in Fig. 4d (Cs, 2A′′). However, the distance between the topmost yttrium atom and the capped oxygen atom is shortened (∼0.216 Å). The structure (Cs, 2A′) (Fig. 4f) was similar to (Cs, 2A′′) (Fig. 4d) in geometrical configuration, which is 0.12 eV less stable. The second low-lying isomer (Cs, 2A′) (Fig. 4e) with three bridging O atoms is only 0.04 eV higher in energy at the BP86 level, which is viewed as the global minimum by Yang et al.58 The corresponding quartet state (D3h, 4A′1) (Fig. 4g) is 0.16 eV above the ground state. As for these low-lying isomers, single-point CCSD(T) are calculated using the BP86 results. The isomer in Fig. 4d is the most stable structure among the above-mentioned four isomers. The other isomers are more than 0.12 eV higher in energy, as given in ESI (Table SI†).
 | ||
| Fig. 5 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3O4−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
The ground state of Y3O4 is predicted to be 2A1 with C3v symmetry (Fig. 5d), which is in agreement with a previous report.59 It is similar to the ground state of Y3O4− cluster in terms of atomic connectivity, except that all the corresponding bond distances become a little shorter. The nearest low-lying isomer (Fig. S5e†) with a terminal and two bridging O atoms is located 0.52 eV above the ground state.
3.3 Stoichiometric clusters: Y3O5−/0
The ground state for Y3O5− is Cs (1A′) (Fig. 6a), in which three Y atoms transfer a total of ten electrons to the oxygen atoms. In the ground state of Y3O5− anionic cluster, the Y–Ot bond distance is 1.926 Å which can be assigned as a typical Y![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond.57 The length of the Y–Oc bond connecting the top Y atom increases to 2.486 Å, compared with 2.175 Å in the ground state of Y3O4− (Fig. 5d). A higher symmetry C2v (1A1) structure is about 0.57 eV above the ground state, as shown in Fig. S6a.†
O double bond.57 The length of the Y–Oc bond connecting the top Y atom increases to 2.486 Å, compared with 2.175 Å in the ground state of Y3O4− (Fig. 5d). A higher symmetry C2v (1A1) structure is about 0.57 eV above the ground state, as shown in Fig. S6a.†
 | ||
| Fig. 6 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3O5−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
The ground state of Y3O5 is predicted to be 2A′′ with Cs symmetry (Fig. 6b). The Y–Ot bond length is 2.155 Å and about 0.229 Å longer compared to that of the anion (Fig. 6a). Nevertheless, the length of the Y–Oc bond connecting the top Y atom becomes a slightly shorter (∼0.177 Å). Other isomers, shown in Fig. 6c and d, are 0.11 eV and 0.21 eV higher, respectively. The results of the single-point CCSD(T) calculations with the BP86 geometries within 0.20 eV indicate that the isomer (Cs, 2A′′) (Fig. 6b) is the ground state and the isomer in Fig. 6c is 0.19 eV above the ground state.
3.4 Oxygen-rich clusters: Y3O6−/0
We started the structural search for the oxygen-rich clusters Y3O6−/0 by adding one oxygen atom (terminal, capped or bridging oxygen atom) from the structures of Y3O5−/0 clusters. The ground state of Y3O6− is the Cs (1A′) (Fig. 7a) structure, which can be viewed as replacing the terminal oxygen in the ground state of Y3O5− (Cs, 1A′) (Fig. 6a) by an O2 unit. The calculated O–O bond length of the O2 unit in the ground state of Y3O6− (Fig. 7a) is 1.492 Å, which is close to that of the free peroxide anion O22− (1.540 Å calculated at the same level). The triplet state isomers (Fig. 7b and e) with an O2 unit are 0.06 eV and 0.27 eV higher in energy. The third low-lying isomer Cs (3A′′) (Fig. 7c) with two terminal oxygen atoms is 0.19 eV higher in energy. Single-point CCSD(T) calculations are performed for the low-lying isomers, revealing that isomer (Cs 1A′) (Fig. 7a) is the global minimum, and is about 0.60 eV lower in energy than isomer (Cs, 3A′′) (Fig. 7b).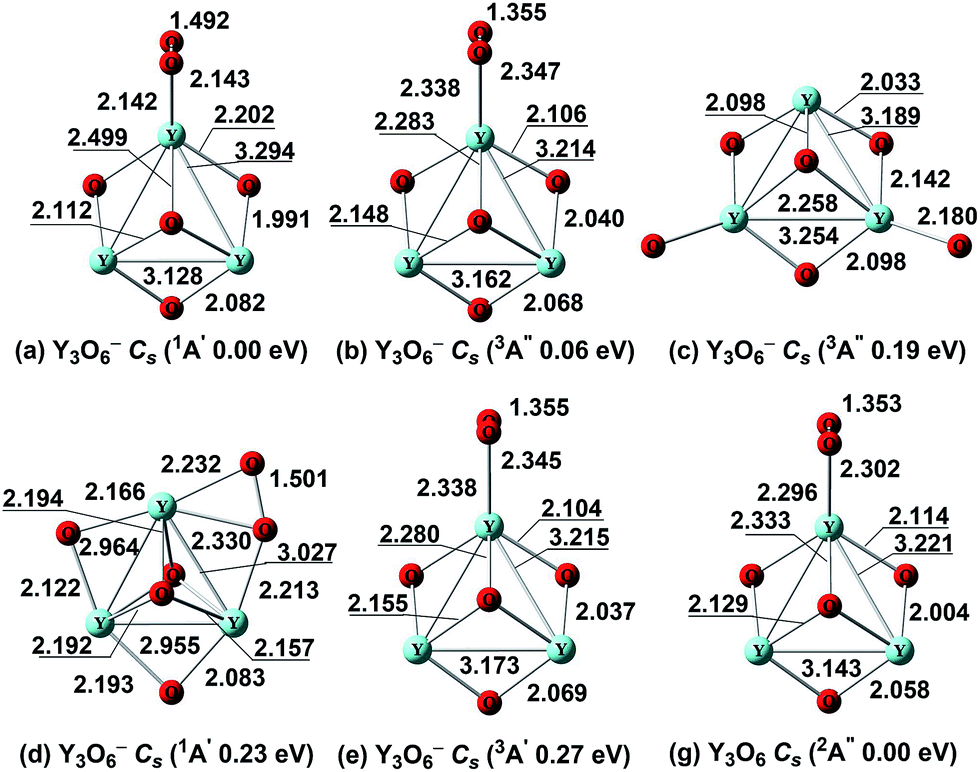 | ||
| Fig. 7 Optimized global minima and selected low-lying structures (within ∼0.40 eV) for Y3O6−/0 clusters at the BP86 level. The bond lengths are in angstroms. | ||
The neutral ground state Cs (2A′′) (Fig. 7g) shows similar structural characteristics relative to its anion, except that the distance of the O2 unit is shortened. The bond length is 1.353 Å for the O2 unit, which is assigned as a superoxide (O2−˙) unit (1.361 Å calculated at the same level). The alternative optimized neutral structures are at least 0.72 eV above the ground state (Fig. S7, ESI†).
4. Discussion
4.1 Interpretation of the simulated photoelectron spectra and molecular orbital analyses
We calculated the vertical detachment energies (VDEs) of Y3Ox− (x = 0–6) on the basis of the identified anionic ground-state structures using the generalized Koopmans' theorem. The calculated VDEs and the simulated PES spectra for the global minima are collected in Table 1 and Fig. 8, respectively. Other VDEs for the selected low-lying isomers are listed in Fig. S8.† All the simulations were done by fitting the distribution of the calculated VDEs with unit-area Gaussian functions of 0.10 eV width. In the single-particle picture, photodetachment involves the removal of electrons from the occupied molecular orbitals (MOs) of an anion. The final states are the ground and excited states of the corresponding neutral. Within the one-electron formalism, each occupied MO for a closed-shell anion will generate a single PES band with the associated vibrational structures governed by the Franck–Condon principle. However, Y3O3− is open-shell with two single unpaired electrons in their lowest-energy structures. In these cases, detachment from a fully occupied MO would result in two detachment channels because of the removal of either the spin-down (α) or the spin-up (β) electrons, giving rise to doublet (D) and quartet (Q) final states, respectively, as given in Table 1. In the following investigation, we will attempt to qualitatively account the simulated PES features using molecular orbital analyses. Considering the complicated nature of the electronic structures of these systems, many of the assignments should be tentatively considered.| Theorya | |||
|---|---|---|---|
| Featureb | MO | VDE | |
| a The labels α and β denote majority and minority spins, whereas D and Q denote doublet and quartet Y3O3 final states upon photodetachment, respectively.b The labels denote the peaks in the simulated PES. | |||
| Y3−, (D3h 1A′) | X | 4a′1 | 1.29 |
| 2a′′2 | 1.32 | ||
| A | 4e | 1.68 | |
| 1.68 | |||
| B | 3a′1 | 3.09 | |
| Y3O−, (C3v 1A1) | X | 7a1 | 1.20 |
| A | 6e | 1.56 | |
| 1.56 | |||
| B | 6a1 | 2.79 | |
| Y3O2−, (Cs 1A′) | X | 17a′ | 1.37 |
| 8a′′ | 1.38 | ||
| A | 16a′ | 2.56 | |
| Y3O3−, (Cs 3A′′) | X | 18a′ (α) | 1.43 (D) |
| A | 12a′′ (α) | 1.69 (D) | |
| B | 17a′ (α); [17a′ (β)] | 2.37 (D); [2.19 (Q)] | |
| Y3O4−, (C3v 1A1) | X | 11a1 | 1.05 |
| A | 10e | 3.92 | |
| 3.92 | |||
| Y3O5−, (Cs 1A′) | X | 24a′ | 2.47 |
| 13a′′ | 2.60 | ||
| 23a′ | 2.77 | ||
| Y3O6−, (Cs 1A′) | X | 14a′′ | 1.97 |
 | ||
| Fig. 8 The simulated photoelectron spectra for the global minima of Y3Ox− (x = 0–6). The simulations are done by fitting the distribution of the calculated vertical detachment energies with unit-area Gaussian functions of 0.10 eV width. | ||
 | ||
| Fig. 9 Pictures of the valence molecular orbitals for the ground states of the Y3Ox− (x = 0–6) clusters. | ||
For Y3O2−, the ground state of Y3O2− is a closed-shell (1A′) Cs (Fig. 3a) configuration with a capped and a bridging O atom. The calculated first VDE for the lowest energy structure Cs (1A′) (Fig. 3a) is 1.37 eV at the BP86 level, which is close to that of 1.60 eV, measured by experiment,14 and the second VDE (1.38 eV) is only 0.01 eV higher than the first VDE. Its valence electronic configuration shown in Fig. 9c is …(16a′)2(8a′′)2(17a′)2. Therefore, photodetachment from HOMO (17a′) and HOMO−1 (8a′′) orbitals yield the first PES band X (Fig. 7c). It is only about 0.17 eV higher than that (1.20 eV) of Y3O− because of the nonbonding (17a′) orbital.
The ground state of Y3O3− is an open shell Cs (3A′′) structure, which can be viewed as adding a bridging O atom on the basis of Y3O2−. Its valence electronic configuration is …(17a′)2(12a′′)1(18a′)1. The first detachment channel for the Y3O3− (Cs, 3A′′) ground state is derived from the orbital 18a′ (α) with the calculated VDE of 1.43 eV, which is in good consistency with the previous experimental value (1.52 eV).14 Fig. 9d presents the molecular orbitals of Y3O3−. The HOMO 18a′ consists of a Y 5s character, which makes the first VDE to slightly increase, in contrast to Y3O− and Y3O2−. The second detachment channel is from 12a′′ (α) with the calculated VDE of 1.69 eV, and the band B is yielded by detaching the electron from the fully occupied 17a′ (α, β) orbital with different VDEs of 2.37 eV and 2.19 eV, respectively.
In the Y3O4− anion, the results of the single-point CCSD(T) calculations manifest that the isomer with a capped and three bridging O atoms is the global minimum. Its valence electron configuration is …(17a1)2(10e)4(18a1)2. The calculated VDE for the band X is 1.05 eV. In Fig. 9e, the HOMO mainly consists of Y 5s character, which is contributed equally by each of the three Y atoms. Similar to the study by Wu,59 each Y atom possesses three valence electrons, and three Y atoms will lose a total of eight electrons to form the Y3O4− cluster. The orbital analysis indicates that the rest of the 5s electrons are distributed by the three yttrium atoms.
4.2 Structural evolution and Sequential oxidation of Y3Ox− (x = 1–6)
The current study shows that the ground states of all Y3Ox− (x = 1–6) clusters are the structures with a capped oxygen atom. Starting from the Y3− triangle structure, the ground state of Y3O− is found to be a pyramid, in which the capped oxygen atom is above the plane of the tri-yttrium equilateral triangle. The next three oxygen atoms successively take up the bridging sites until Y3O4−, which is based on a six-membered Y3O3 ring, adds a capped oxygen atom. On the basis of the ground state of Y3O4−, the subsequently added oxygen atom is shown to occupy the terminal site, forming Y3O5−, and the ground state of Y3O6− can be viewed as replacing a terminal O atom in Y3O5− by an O2 unit. In Fig. 10, the trend of electron affinities (EAs) is consistent with that of the first VDEs until Y3O4, which indicates the corresponding anionic and neutral clusters Y3Ox−/0 (x = 1–4), possess the similar geometric configurations. | ||
| Fig. 10 The first vertical detachment energies and electron affinities for the ground states Y3Ox− (x = 1–6) clusters as a function of O content. | ||
Fig. 10 displays the trend of the first VDEs for Y3Ox− (x = 1–6). The first VDEs of the Y3Ox− (x = 1–4) oxide clusters are all above 1.0 eV, 1.20 eV for Y3O−, 1.37 eV for Y3O2−, 1.43 eV for Y3O3−, and 1.05 eV for Y3O4−. For Y3O5−, the first VDE increases to 2.47 eV, while the first VDE for Y3O6− decreases to 1.97 eV. Y has an electron configuration of 4d15s2, and the bare tri-yttrium anion Y3− has 10 valence electrons. The Y-derived valence electrons sequentially transfer to the added oxygen atoms with increasing x. The values of the first VDEs for Y3Ox− (x = 1–4) slightly change as a function of O content. The higher binding energy for Y3O5− can be explained by the nature of the HOMO (24a′) (Fig. 9f), which is primarily the terminal oxygen atom 2p-based orbital. The first VDE for Y3O6− decreases compared with that for Y3O5− because of the electrons derived from π* molecular orbital. The trend of VDEs in Y3Ox− (x = 1–6) is consistent with the previous studies of metal oxide clusters,23,60–64 suggesting the sequential oxidation of the metal oxides clusters.
4.3 All-metal aromaticity of Y3−
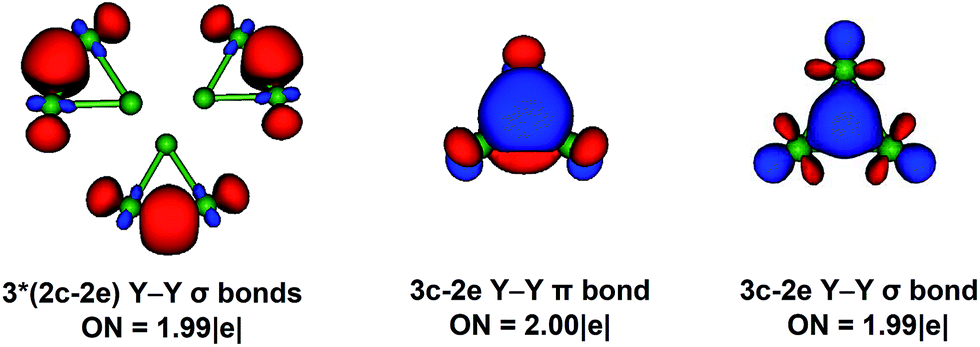 | ||
| Fig. 11 Chemical-bonding elements revealed by the AdNDP analysis of the Y3− cluster. The ONs are reported at the BP86/lanl2dz level of theory. | ||
4.4 Y3O4−/0 as reduced molecular models for dioxygen activation: Y3O4−/0 + O2 → Y3O6−/0
As mentioned earlier, the ground states of Y3O6 (Fig. 7g) and Y3O6− (Fig. 7a) clusters can be viewed as addition of a superoxide O2−˙ and peroxide O22− units on Y3O4 and Y3O4− clusters, respectively. The observation of O2 units in Y3O6 and Y3O6− provides us with an excellent opportunity to study the activation of O2 by the Y3O4 and Y3O4− clusters. The energies of the O2 addition reactions are evaluated at the BP86 level:| Y3O4 (C3v, 2A1) + O2 → Y3O6 (Cs, 2A′′) − 2.97 eV | (1) |
| Y3O4− (C3v, 1A1) + O2 → Y3O6− (Cs, 1A′) − 3.25 eV | (2) |
Eqn (1) and (2) can be viewed as the reactions of O2 with the ground states of Y3O4 and Y3O4−, respectively. Our calculation yielded the O2 chemisorption energy of −2.97 eV in Y3O4 and −3.25 eV in Y3O4−. Similar to our previous studies of metal oxide clusters,22,66–68 molecular orbitals in the O-rich systems are analyzed to obtain further insight into the nature of the chemical bonding. The orbitals of Y3O6 are illustrated in Fig. 12. The singly occupied MO is one of the two π* orbitals of the bound O2 unit, and the HOMO are the doubly occupied π* orbital of O2 that is singly occupied in free O2. The O–O distance (1.353 Å) in Y3O6 (Fig. 6g) is longer than that in free O2 and is close to that in the superoxide O2−˙ unit. It shows that the rest of the 5s electrons of Y3O4 may be readily transferred to the π* orbitals of an approaching O2 molecule, making Y3O4 a reductive agent for O2 activation. Analogously, the O–O distance (1.492 Å) in Y3O6− (Fig. 7a) is close to that in the peroxide O22− unit and the extra two 5s electrons of Y3O4− may be transferred to the π* orbitals of an approaching O2 molecule. Therefore, the Y3O6 and Y3O6− clusters can be considered to be the oxidation products for the activation of O2 through electron transfer between the reduced metal site and dioxygen at the metal oxide surface.
 | ||
| Fig. 12 Molecular orbital pictures of the two highest-energy occupied orbitals of Y3O6. | ||
5. Conclusion
We report a systematic photoelectron spectroscopy and density functional study of a series of tri-nuclear yttrium oxide clusters: Y3Ox− and Y3Ox (x = 0–6). Extensive DFT calculations are performed at the BP86 level to locate the ground states for the Y3Ox−/0 (x = 0–6) cluster. The ground states for Y3− and Y3 are D3h (1A′) and D3h (2A′1) structures, respectively. The tri-yttrium oxide clusters Y3Ox−/0 (x = 1–6) all have a capped oxygen atom. Starting from Y3O−, the successive addition of the bridging oxygen atoms, yield the ground states of Y3O2−, Y3O3− and Y3O4−. Nevertheless, the next added oxygen atom is found to be a terminal one in the Y3O5− cluster and the oxygen-rich cluster Y3O6− is found to have an O2 unit. The neutral species have similar structures with their corresponding anionic clusters. As for the oxygen-rich clusters Y3O6− and Y3O6, O22− and O2−˙ units are found, which can be viewed as Y3O4− and Y3O4 that are oxidized by O2, respectively. Molecular orbital analyses are carried out to elucidate the chemical bonding of these clusters and provide insights into the sequential oxidation from Y3O− to Y3O6−. The molecular orbital analysis presents that Y3− (D3h, 1A′1) possesses σ- and π-aromaticity. The AdNDP analysis reveals that one 3c–2e σ-bond and one 3c–2e π-bond completely delocalized over the three yttrium atoms, which further proves the characteristic of aromaticity in the Y3− cluster.Acknowledgements
We gratefully acknowledge supports from the National Natural Science Foundation of China (21071031, 21301030, 21371034 and 21373048), and the Natural Science Foundation of Fujian Province for Distinguished Young Investigator Grant (2013J06004).Reference
- D. Kima, S. Jeonga, J. Moona and S. H. Cho, J. Colloid Interface Sci., 2006, 297, 589 CrossRef PubMed.
- P. V. A. Padmanabhan, S. Ramanathan, K. P. Sreekumar, R. U. Satpute, T. R. G. Kutty, M. R. Gonal and L. M. Gantayet, Mater. Chem. Phys., 2007, 106, 416 CrossRef CAS PubMed.
- A. M. Pires, O. A. Serra and M. R. Davolos, J. Lumin., 2005, 113, 174 CrossRef CAS PubMed.
- W. J. Song, J. S. Li, T. B. Zhang, H. C. Kou and X. Y. Xue, J. Power Sources, 2014, 245, 808 CrossRef CAS PubMed.
- Y. Chen, J. Bunch, C. Jin, C. H. Yang and F. L. Chen, J. Power Sources, 2012, 204, 40 CrossRef CAS PubMed.
- V. Singh, V. K. Rai, B. Voss, M. Haase, R. P. S. Chakradhar, D. T. Naidu and S. H. Kim, Spectrochim. Acta, Part A, 2013, 109, 206 Search PubMed.
- A. M. Pires, O. A. Serra and M. R. Davolos, J. Alloys Compd., 2004, 374, 181 CrossRef CAS PubMed.
- N. Al-Yassir and R. L. V. Mao, Can. J. Chem., 2008, 86, 146 CrossRef CAS.
- N. Al-Yassir and R. L. V. Mao, Appl. Catal., A, 2007, 317, 275 CrossRef CAS PubMed.
- J. B. Ma, Z. C. Wang, M. Schlangen, S. G. He and H. Schwarz, Angew. Chem., Int. Ed., 2013, 52, 1226 CrossRef CAS PubMed.
- V. V. Lavrov, V. Blagojevic, G. K. Koyanagi, G. Orlova and D. K. Bohme, J. Phys. Chem. A, 2004, 108, 5610 CrossRef CAS.
- M. Knickelbein, Chem. Phys., 1995, 102, 1 CAS.
- H. B. Wu and L. S. Wang, J. Phys. Chem. A, 1998, 102, 9129 CrossRef CAS.
- A. Pramann, Y. Nakamura and A. Nakajima, J. Phys. Chem. A, 2001, 105, 7534 CrossRef CAS.
- G. Gu, B. Dai, X. Ding and J. Yang, Eur. Phys. J. D, 2004, 29, 27 CrossRef CAS.
- B. Dai, K. Deng and J. Yang, Chem. Phys. Lett., 2002, 364, 188 CrossRef CAS.
- Z. Yang and S. J. Xiong, J. Chem. Phys., 2008, 129, 124308 CrossRef PubMed.
- A. B. Rahane, P. A. Murkute, M. D. Deshpande and V. Kumar, J. Phys. Chem. A, 2013, 117, 5542 CrossRef CAS PubMed.
- H. J. Zhai, X. Huang, T. Waters, X. B. Wang, R. A. J. O'Hair, A. G. Wedd and L. S. Wang, J. Phys. Chem. A, 2005, 109, 10512 CrossRef CAS PubMed.
- H. J. Zhai, X. Huang, L. F. Cui, X. Li, J. Li and L. S. Wang, J. Phys. Chem. A, 2005, 109, 6019 CrossRef CAS PubMed.
- H. J. Zhai, B. Wang, X. Huang and L. S. Wang, J. Phys. Chem. A, 2009, 113, 9804 CrossRef CAS PubMed.
- B. Wang, W. J. Chen, B. C. Zhao, Y. F. Zhang and X. Huang, J. Phys. Chem. A, 2010, 114, 1964 CrossRef CAS PubMed.
- W. J. Chen, H. J. Zhai, Y. F. Zhang, X. Huang and L. S. Wang, J. Phys. Chem. A, 2010, 114, 5958 CrossRef CAS PubMed.
- Q. Zhou, W. C. Gong, L. Xie, C. G. Zheng, W. Zhang, B. Wang, Y. F. Zhang and X. Huang, Spectrochim. Acta, Part A, 2014, 117, 651 CrossRef CAS PubMed.
- B. Wang, H. J. Zhai, X. Huang and L. S. Wang, J. Phys. Chem. A, 2008, 112, 10962 CrossRef CAS PubMed.
- P. W. Fowler, R. W. A. Havenith and E. Steiner, Chem. Phys. Lett., 2002, 359, 530 CrossRef CAS.
- B. B. Averkiev and A. I. Boldyrev, J. Phys. Chem. A, 2007, 111, 12864 CrossRef CAS PubMed.
- T. R. Galeev and A. I. Boldyrev, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2011, 107, 124 RSC.
- A. E. Kuznetsov, J. D. Corbett, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2001, 40, 3369 CrossRef CAS.
- Z. Badri, S. Pathak, H. Fliegl, P. Rashidi-Ranjbar, R. Bast, R. Marek, C. Foroutan-Nejad and K. Ruud, J. Chem. Theory Comput., 2013, 9, 4789 CrossRef CAS.
- X. X. Chi and Y. Liu, Int. J. Quantum Chem., 2007, 107, 1886 CrossRef CAS.
- F. Feixas, E. Matito, M. Duran, J. Poater and M. Sol, Theor. Chem. Acc., 2011, 128, 419 CrossRef CAS PubMed.
- A. E. Kuznetsov, K. A. Birch, A. I. Boldyrev, X. Li, H. J. Zhai and L. S. Wang, Science, 2003, 300, 622 CrossRef CAS PubMed.
- H. J. Zhai, W. J. Chen, X. Huang and L. S. Wang, RSC Adv., 2012, 2, 2707 RSC.
- X. Huang, H. J. Zhai, B. Kiran and L. S. Wang, Angew. Chem., Int. Ed., 2005, 44, 7251 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, Pseudopotentials of the Stuttgart/Dresden group 1998, revision, August 11, 1998, http://www.theochem.uni-stuttgart.de/pseudopotentiale.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS PubMed.
- D. J. Tozer and N. C. Handy, J. Chem. Phys., 1998, 109, 10180 CrossRef CAS PubMed.
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910 CrossRef CAS PubMed.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer III, J. Chem. Phys., 1988, 89, 7382 CrossRef CAS PubMed.
- K. Raghavachari and G. W. Trucks, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718 CrossRef CAS PubMed.
- R. J. Bartlett and M. Musiał, Rev. Mod. Phys., 2007, 79, 291 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylow, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H. J. Werner, P. J. Knowles, F. R. Manby, M. Schütz, P. Celani, G. Knizia, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. HamPel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. KöPPl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version 2010.1, a package of ab initio programs, http://www.molpro.net Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS.
- L. Andrews, M. F. Zhou and G. V. Chertihin, J. Phys. Chem. A, 1999, 103, 6525 CrossRef CAS.
- Y. Gong, C. F. Ding and M. F. Zhou, J. Phys. Chem. A, 2009, 113, 8569 CrossRef CAS PubMed.
- D. Yu. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC.
- U. Varetto, Molekel5.4, Swiss National Supercomputing Centre, Manno, Switzerland, 2009 Search PubMed.
- D. Dai and K. Balasubramanian, J. Chem. Phys., 1993, 98, 7098 CrossRef CAS PubMed.
- Y. X. Zhao, X. L. Ding, Y. P. Ma, Z. C. Wang and S. G. He, Theor. Chem. Acc., 2010, 127, 449 CrossRef CAS PubMed.
- Z. Yang and S. J. Xiong, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 245101 CrossRef.
- L. Wu, C. Zhang, S. A. Krasnokutski and D. S. Yang, J. Chem. Phys., 2012, 137, 084312 CrossRef PubMed.
- H. Wu, X. Li, X. B. Wang, C. F. Ding and L. S. Wang, J. Chem. Phys., 1998, 109, 449 CrossRef CAS PubMed.
- L. S. Wang, H. Wu and S. R. Desai, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 53, 8028 CrossRef.
- L. S. Wang, H. Wu and S. R. Desai, Phys. Rev. Lett., 1996, 76, 4853 CrossRef CAS.
- S. J. Lin, X. H. Zhang, L. Xu, B. Wang, Y. F. Zhang and X. Huang, J. Phys. Chem. A, 2013, 117, 3093 CrossRef CAS PubMed.
- L. F. Wang, L. Xie, H. L. Fang, Y. F. Li, X. B. Zhang, B. Wang, Y. F. Zhang and X. Huang, Spectrochim. Acta, Part A, 2014, 131, 446 CrossRef CAS PubMed.
- A. P. Sergeeva, B. B. Averkiev and A. I. Boldyrev, Struct. Bonding, 2010, 136, 275 CrossRef CAS.
- H. J. Zhai, X. H. Zhang, W. J. Chen, X. Huang and L. S. Wang, J. Am. Chem. Soc., 2011, 133, 3085 CrossRef CAS PubMed.
- X. Huang, H. J. Zhai, T. Waters, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2006, 45, 657 CrossRef CAS PubMed.
- S. J. Lin, W. C. Gong, L. F. Wang, W. B. Liu, B. C. Zhao, B. Wang, Y. F. Zhang and X. Huang, Theor. Chem. Acc., 2014, 133, 1435 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09202c |
| This journal is © The Royal Society of Chemistry 2014 |