
An efficient synthesis of novel dibenzoxdiazepine-fused heterocycles through a multicomponent reaction†
Zhiwei Qian,
Anjiang Yang,
Weiteng An,
Ting Yu,
Xin Wang,
Yongliang Zhang,
Jingkang Shen* and
Tao Meng*
State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai 201203, PR China. E-mail: jkshen@mail.shcnc.ac.cn; tmeng@sibs.ac.cn
First published on 7th October 2014
Abstract
A one-pot synthesis of 6-oxa-2,2a1,11-triazadibenzo[cd,g]azulenes by a three-component reaction of a 2-aminoheterocycle, aldehydes, and 2-isocyanophenyl acetate is presented. This efficient and green protocol has the advantages of environmental friendliness, high yields and operational simplicity. The atropisomeric properties of this unique structure were examined by 1H NMR spectroscopy and X-ray structural analyses, and the barriers to their interconversion were clarified.
Polycyclic heterocycles are frequently found in natural products and pharmaceutical agents. Among them, functionalized dibenzoxazepines are a very important class of seven-membered rings and such structural units could widely exist in numerous medicinal heterocyclic molecules with promising biological and pharmaceutical activities.1 These biological characteristics have stimulated organic researchers to explore synthetic methods for dibenzoxazepines and their structural analogues.2 Recently, isocyanide-based multicomponent reactions (IMCR) followed by other synthetic transformations have emerged as a powerful tool for creating fused multicyclic skeletons. As a part of our program3 to discover novel heterocycles as antitumor agents based on imidazo[1,2-a]pyridine ring system, which may be regarded as a privileged structure.4 Following this strategy, our next challenge was the introduction of an additional benzoxdiazepine ring in the imidazo[1,2-a]pyridine framework to form the 6-oxa-2,2a,1 11-triazadibenzo[cd,g]azulene as shown in Scheme 1. We envisaged that this scaffold might be synthesized from halogen intermediate A, which itself could be prepared from 6-bromopyridin-2-amine, an aldehyde and 2-isocyanophenol via a Groebke–Blackburn–Bienaymé (GBB) reaction,5 followed by in situ intramolecular cyclization to afford this polycyclic heterocycle (Scheme 1). To the best of our knowledge, this series of compounds has never been reported.
 | ||
| Scheme 1 Retro-synthetic approach for substituted 6-oxa-2,2a,1 11-triazadibenzo[cd,g]azulene. | ||
To test the hypothesis, we commenced the investigation with 6-bromopyridin-2-amine 1a (0.2 mmol), benzaldehyde 2a (0.2 mmol) and 2-isocyanophenyl acetate 3 (0.2 mmol)6 as model substrates. The desired GBB condensation intermediate 4a was afforded in 91% yield using catalyst-free and solvent-free conditions, which was recently developed by Sharada et al.7 Subsequently, substrate 4a was used for the optimization of the cyclization reaction conditions, including different catalysts and various solvents, and the results are summarized in Table 1. No reaction occurred in the absence of base (entry 1, Table 1). We envisaged that the acetyl group might be removed under basic condition. Various bases (entries 2–6, Table 1) were used in the intramolecular cyclization, however, we found that the acetyl group was easily transferred to the adjacent nitrogen to form the acetylamide 5a. No deacetylated product 5a′ was observed in the reaction process. To our delight, the isolated yield of 5a was enhanced further to 83% when the potassium carbonate was used as the base (entry 3, Table 1). Using weaker bases, such as sodium carbonate (entry 2, Table 1), sodium bicarbonate (entry 4, Table 1), potassium acetate (entry 5, Table 1), or stronger bases, such as sodium hydroxide (entry 6, Table 1) afford the product in lower yields compare to potassium carbonate. Moreover, no obvious improvement in the yield was observed when the solvent was switched to isopropanol and DMF (entries 7 and 8, Table 1). Next, typical Ullmann and Buchwald–Hartwig cross coupling reaction conditions were used and no significant improvement in the yield (entries 9 and 10, Table 1). The structure of 5a was unambiguously established by X-ray crystallographic analysis (Fig. 1).
|
||||
|---|---|---|---|---|
| Entry | Base | Additive | Solvent | Yieldb (%) |
| a Reaction conditions: (i) 1a (1 equiv.), 2a (1.1 equiv.), 3 (1.2 equiv.), 140 °C, 10 min, 90% yield; (ii) base (2.0 equiv.), 100 °C, 2 h.b Isolated yield of step ii. | ||||
| 1 | None | None | Dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O(4:1) :H2O(4:1) |
0 |
| 2 | Na2CO3 | None | Dioxane:H2O(4:1) |
63 |
| 3 | K2CO3 | None | Dioxane:H2O(4:1) |
83 |
| 4 | NaHCO3 | None | Dioxane:H2O(4:1) |
31 |
| 5 | KOAc | None | Dioxane:H2O(4:1) |
40 |
| 6 | NaOH | None | Dioxane:H2O(4:1) |
15 |
| 7 | K2CO3 | None | Isopropanol, reflux | 70 |
| 8 | K2CO3 | None | DMF | 78 |
| 9 | K2CO3 | CuI/TMEDA | Dioxane:H2O(4:1) |
70 |
| 10 | K2CO3 | Pd(OAc)2/XPhos | Toluene | 62 |

 | ||
| Fig. 1 X-ray structure of compound 5a.8 | ||
On the basis of the results obtained above, a plausible mechanism of this reaction is illustrated in Scheme 2. First, the reaction is expected to proceed via the in situ formation of A. The next step involves aminolysis of acetylphenol to form intermediate C. Finally, after intramolecular cyclization of C to afford the final product D.
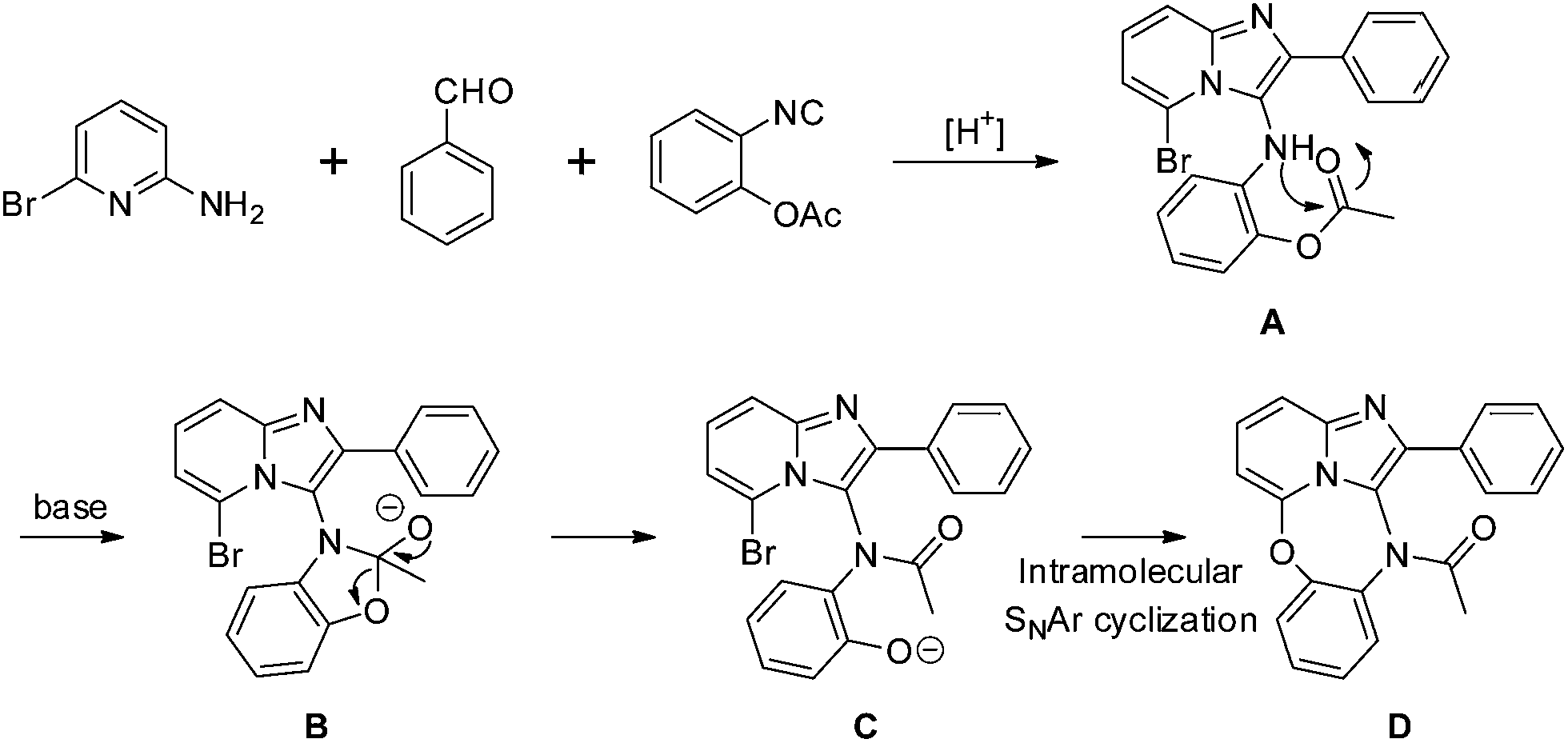 | ||
| Scheme 2 The possible reaction mechanism. | ||
With the optimal conditions established, we then investigated the scope of this method. The simplicity of a one-pot procedure is perfectly amenable to automation for combinatorial synthesis. Likewise, all the syntheses were performed on a parallel synthesizer (Radleys Discovery Technology, Carousel 12 Place Reaction Station) to give corresponding products 5a–t (Table 2). First, we examined the reactions of various aldehydes 2a–q with 6-bromopyridin-2-amine (1a) and 2-isocyanophenyl acetate (3), which proceeded smoothly and efficiently to produce the corresponding products (5a–q) in yields ranging from 36 to 65%. Moreover, we were pleased to find that the pyridine core could be successfully extended to pyrazine. For example, 6-chloropyrazin-2-amine (1b) smoothly reacted with benzaldehydes (2a–c) and 2-isocyanophenyl acetate (3) to give the expected product 5r–t in around 42% yields (Table 2, entries 18–20).
|
|||
|---|---|---|---|
| Entry | Cpds | R | Yieldb (%) |
| a Reaction conditions: 1a, 1b (1.0 mmol, 1.0 equiv.), aldehydes 2a–q (1.05 equiv.), 2-isocyanophenyl acetate 3 (1.05 equiv.), 140 °C, 10 min then dissolved in dioxane:H2O(4:1), K2CO3 (2.0 equiv.), 100 °C, 1 h.b Isolated yields. |
|||
| 1 | 5a | Ph (2a) | 55% |
| 2 | 5b | 4-MeO-Ph (2b) | 58% |
| 3 | 5c | 4-CF3-Ph (2c) | 62% |
| 4 | 5d | 4-Me-Ph (2d) | 56% |
| 5 | 5e | 4-CN-Ph (2e) | 65% |
| 6 | 5f | 4-Br-Ph (2f) | 65% |
| 7 | 5g | 4-[1,1′-biphenyl] (2g) | 51% |
| 8 | 5h | 2-Br-Ph (2h) | 50% |
| 9 | 5i | 2-MeO-Ph (2i) | 49% |
| 10 | 5j | 3,5-Difluor-Ph (2j) | 49% |
| 11 | 5k | 2-Pyrrole (2k) | 50% |
| 12 | 5l | 2-Thiophene (2l) | 52% |
| 13 | 5m | 2-Furan (2m) | 48% |
| 14 | 5n | 2-Pyridine (2n) | 36% |
| 15 | 5o | 3-Pyridine (2o) | 40% |
| 16 | 5p | Cyclohexane (2p) | 36% |
| 17 | 5q | n-Propyl (2q) | 56% |
| 18 | 5r | Ph (2a) | 40% |
| 19 | 5s | 4-MeO-Ph (2b) | 44% |
| 20 | 5t | 4-CF3-Ph (2c) | 42% |

While measuring the 1H-NMR, we observed that compounds bearing the EWG substituted phenyl ring as the R substitution showed more complicated NMR signals compare to the EDG substituted compounds. We assumed this effect might be due to the atropisomer effect, which is very common for amide type of compound due to the pyramidal inversion. A variable-temperature 400 MHz 1H NMR spectra in d6-DMSO study of 5c was carried out in order to verify the co-existence of two conformers (see ESI†). The acetyl dibenzoxdiazepine thus obtained were expected to exist as racemates of the atropisomers due to the axial chirality at aryl-N(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).9 On the basis of its X-ray analysis, 5cA was assigned to be (aR), and hence, 5cB to be (aS) (Fig. 2).7 We managed to obtain each enantiomer of 5c using preparative chiral HPLC, however, racemization occurred immediately after separation, in the end we could only obtain each enantiomers at about 50% ee. We examined the stereochemical stability of the enantiomers (5cA and 5cB) and found that it was estimated to be low: racemization occurred after storage for 2 h at 25 °C in EtOH. The activation free-energy barrier to rotation (ΔG‡) was measured and calculated to be 97.5 kJ mol−1 (see ESI†).10
O).9 On the basis of its X-ray analysis, 5cA was assigned to be (aR), and hence, 5cB to be (aS) (Fig. 2).7 We managed to obtain each enantiomer of 5c using preparative chiral HPLC, however, racemization occurred immediately after separation, in the end we could only obtain each enantiomers at about 50% ee. We examined the stereochemical stability of the enantiomers (5cA and 5cB) and found that it was estimated to be low: racemization occurred after storage for 2 h at 25 °C in EtOH. The activation free-energy barrier to rotation (ΔG‡) was measured and calculated to be 97.5 kJ mol−1 (see ESI†).10
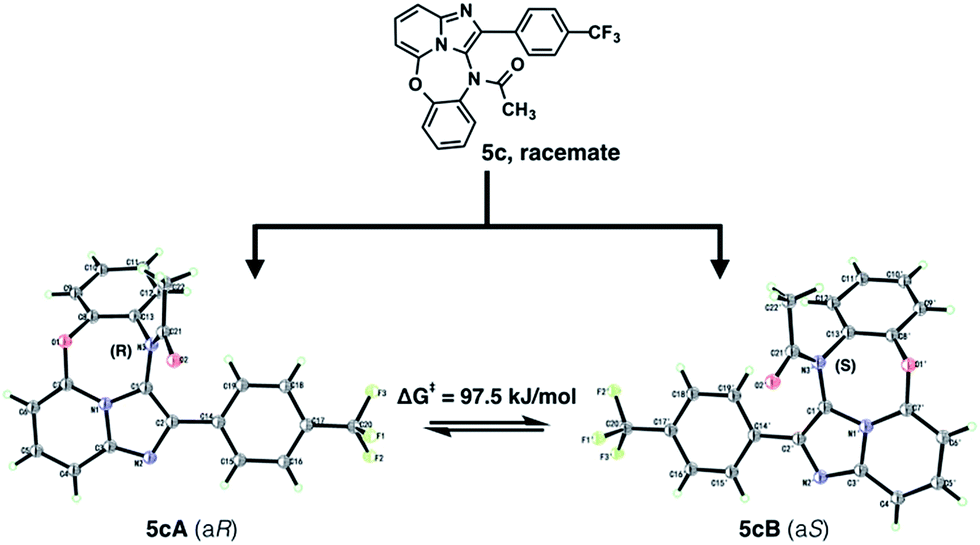 | ||
| Fig. 2 X-ray analysis of atropisomer 5c. | ||
In summary, we have developed a clean and efficient method for the sequential synthesis of new functionalized 6-oxa-2,2a,1 11-triazadibenzo[cd,g]azulene derivatives, which were characterized by means of 1H NMR, 13C NMR, HRMS and X-ray. Easily available starting materials, metal catalyst-free conditions and good yields are the main advantages of this method. We hope that these stereochemical findings in acetyl dibenzoxazepine will assist in future drug design in which heterocyclic systems are utilized as the core structure for biologically active molecules.
Acknowledgements
This work was supported by the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (no.: 2012ZX09103-101-026) and the National Natural Science Foundation of China (grant 81473130, 81001355 and 81273367).Notes and references
- (a) M. M. Mc Gee, S. Gemma, S. Butini, A. Ramunno, D. M. Zisterer, C. Fattorusso, B. Catalanotti, G. Kukreja, I. Fiorini, C. Pisano, C. Cucco, E. Novellino, V. Nacci, D. C. Williams and G. Campiani, J. Med. Chem., 2005, 4367 CrossRef CAS PubMed; (b) H. J. M. Gijsen, D. Berthelot, M. Zaja, B. Brone, I. Geuens and M. Mercken, J. Med. Chem., 2010, 53, 7011 CrossRef CAS PubMed; (c) K. Kubota, H. Kurebayashi, H. Miyachi, M. Tobe, M. Onishi and Y. Isobe, Bioorg. Med. Chem., 2011, 19, 3005 CrossRef CAS PubMed; (d) G. Campiani, V. Nacci, I. Fiorini, M. P. De Filippis, A. Garofalo, S. M. Ciani, G. Greco, E. Novellino, D. C. Williams, D. M. Zisterer, M. J. Woods, G. Mihai, C. Manzoni and T. Mennini, J. Med. Chem., 1996, 39, 3435 CrossRef CAS PubMed; (e) T. Miki, M. Kori, H. Mabuchi, H. Banno, R. Tozawa, M. Nakamura, S. Itokawa, Y. Sugiyama and H. Yukimasa, Bioorg. Med. Chem., 2002, 10, 401 CrossRef CAS.
- (a) H. S. Lim, Y. L. Choi and J. N. Heo, Org. Lett., 2013, 15, 4718 CrossRef CAS PubMed; (b) P. Sang, M. Yu, H. F. Tu, J. W. Zou and Y. H. Zhang, Chem. Commun., 2013, 49, 701 RSC; (c) E. A. El-Rady, J. Heterocycl. Chem., 2002, 39, 1109 CrossRef CAS; (d) M. Ghafarzadeh, E. S. Moghadam and F. Faraji, J. Heterocycl. Chem., 2013, 50, 754 CrossRef CAS; (e) Y. C. Lin, N. C. Li and Y. J. Cherng, J. Heterocycl. Chem., 2014, 51, 808 CrossRef CAS.
- (a) H. Y. Zhou, W. Wang, O. Khorev, Y. L. Zhang, Z. H. Miao, T. Meng and J. K. Shen, Eur. J. Org. Chem., 2012, 5585 CrossRef CAS; (b) A. J. Yang, R. W. Jiang, O. Khorev, T. Yu, Y. L. Zhang, L. P. Ma, G. H. Chen, J. K. Shen and T. Meng, Adv. Synth. Catal., 2013, 355, 1984 CrossRef CAS; (c) H. P. Sun, H. Y. Zhou, O. Khorev, R. W. Jiang, T. Yu, X. Wang, Y. L. Du, Y. Ma, T. Meng and J. K. Shen, J. Org. Chem., 2012, 77, 10745 CrossRef CAS PubMed.
- (a) A. M. Palmer, B. Grobbel, C. Jecke, C. Brehm, P. J. Zimmermann, W. Buhr, M. P. Feth, W. A. Simon and W. J. Kormer, Med. Chem., 2007, 50, 6240 CrossRef CAS PubMed; (b) M. A. Ismail, R. Brun, T. Wenzler, F. A. Tanious, W. D. Wilson and D. W. Boykin, J. Med. Chem., 2004, 47, 3658 CrossRef CAS PubMed.
- (a) H. Bienayme and K. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234 CrossRef CAS; (b) C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635 CrossRef CAS.
- (a) M. C. Pirrung and S. Ghorai, J. Am. Chem. Soc., 2006, 128, 11772 CrossRef CAS PubMed; (b) M. C. Pirrung, S. Ghorai and T. R. Ibarra-Rivera, J. Org. Chem., 2009, 74, 4110 CrossRef CAS PubMed.
- S. Vidyacharan, A. H. Shinde, B. Satpathi and D. S. Sharada, Green Chem., 2014, 16, 1168 RSC.
- CCDC-1007734 (5a) and CCDC-1007733 (5c) contain the supplementary crystallographic data for this paper†.
- (a) H. Tabata, T. Yoneda, T. Oshitari, H. Takahashi and H. Natsugari, J. Org. Chem., 2013, 78, 6264 CrossRef CAS PubMed; (b) H. Tabata, N. Wada, Y. Takada, T. Oshitari, H. Takahashi and H. Natsugari, J. Org. Chem., 2011, 76, 5123 CrossRef CAS PubMed; (c) H. Tabata, N. Wada, Y. Takada, J. Nakagomi, T. Miike, H. Shirahase, T. Oshitari, H. Takahashi and H. Natsugari, Chem.–Eur. J., 2012, 18, 1572 CrossRef CAS PubMed; (d) K. Akiba, H. Tabata, S. Lee, H. Takahashi and H. Natsugari, Org. Lett., 2008, 10, 4871 CrossRef PubMed; (e) H. Tabata, H. Suzuki, K. Akiba, H. Takahashi and H. Natsugari, J. Org. Chem., 2010, 75, 5984 CrossRef CAS PubMed.
- M. Petit, A. J. B. Lapierre and D. P. Curran, J. Am. Chem. Soc., 2005, 127, 14994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, compound characterization data, 1H and 13C NMR spectra of new compounds. CCDC [1007733, 1007734]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra09196e |
| This journal is © The Royal Society of Chemistry 2014 |