
Enhanced supercapacitor performance by incorporating nickel in manganese oxide†
Abstract
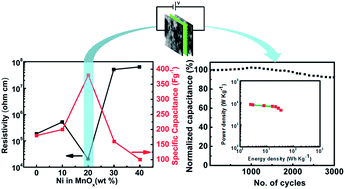
Nickel manganese mixed oxides (NiyMn1−yOx; 0 ≤ y ≤ 0.4) have been synthesized by in situ inclusion of nickel during the growth of manganese oxide (MnOx). The effect of nickel concentration in MnOx is investigated by cyclic voltammetry, current–voltage characteristics, scanning electron microscopy and N2 adsorption–desorption analysis. Variations in electronic conductivity and specific capacitance suggest that nickel concentration in the MnOx matrix significantly affects the supercapacitor electrode performance. At Ni/Mn ∼0.25, i.e. Ni0.2Mn0.8Ox, the material crystallizes into spinel NiMn2O4 as a prominent phase and exhibits a specific surface area (118 m2 g−1) with a granular morphology. Furthermore Ni0.2Mn0.8Ox exhibited low resistivity (2.07 × 104 Ohm cm) and consequently high specific capacitance ∼380 F g−1, endowing additional merits. The fabricated supercapacitor device (Ni0.2Mn0.8Ox//Ni0.2Mn0.8Ox) delivers 35 W h kg−1 energy density and 3.74 kW kg−1 power density with remarkably high capacitive retention ∼92% after 3000 galvanostatic charge–discharge cycles. These encouraging results show great potential in developing energy storage devices from manganese oxide based electrodes incorporating nickel in the lattice.
 Please wait while we load your content...
Please wait while we load your content...

