
Effects of ultrasonic cavitation intensity on the efficient liquid-exfoliation of MoS2 nanosheets
Abstract
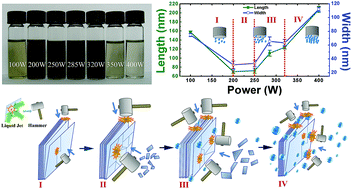
Liquid exfoliation has been widely used to yield two dimensional layered materials in laboratory because of its simplicity and ease for mass production characteristics. In this study, the dispersions and morphology of exfoliated MoS2 in N-methyl-2-pyrrolidone (NMP) solvent at different ultrasonic powers are investigated. An optimal power to exfoliate MoS2 nanoflakes in NMP for high yield and small lateral size with narrow size distribution is obtained. Our results showed that the concentration of dispersions did not monotonously increase with growing ultrasonic power, but rather initially increased with input power, and then decreased after 320 W due to the cavitation shielding effect. The flake size decreased with ultrasonic power from 100 W to 250 W and then slightly increased; after 320 W, the average lateral size of flakes dramatically increased and a wide size distribution with relatively large scale nano-flakes was detected. The mechanism of ultrasonic cavitation effect on the concentration and morphology has been analyzed.
 Please wait while we load your content...
Please wait while we load your content...

