
Glucose oxidase-encapsulated nanogold hollow microspheres as labels based on a sensitive electroluminescent immunoassay
Xiu Wanga,
Zhaoquan Biana,
Chengchao Chua,
Xiaoxiao Zhenga,
Shenguang Geb,
Jinghua Yu a,
Mei Yan*a and
Xianrang Song*c
a,
Mei Yan*a and
Xianrang Song*c
aKey Laboratory of Chemical Sensing & Analysis in Universities of Shandong, School of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022, P. R. China. E-mail: chm_yanm@126.com; Fax: +86-531-88959988; Tel: +86-531-82767161
bShandong Provincial Key Laboratory of Preparation and Measurement of Building Materials, University of Jinan, Jinan 250022, China
cCancer Research Center, Shandong Tumor Hospital, Jinan 250012, P. R. China
First published on 1st October 2014
Abstract
We herein report the synthesis of glucose oxidase-encapsulated nanogold hollow microspheres (GOx–GHS), which show excellent catalytic performance for the reduction reaction of glucose, and the nanogold on the GOx–GHS provided the nanoparticles with good biocompatibility. In addition, the electrochemiluminescence (ECL) property of luminol was investigated and a type of luminol nano-composite (AuNPs–EGN–luminol) was prepared. The as-prepared nano-composite was found to have good ECL property of the luminol, as well as good electroconductibility. The prepared GOx–GHS and AuNPs–EGN–luminol were applied in a sandwich-type ECL immunosensor for cancer antigen 125 (CA125). In the immunosensor, AuNPs–EGN–luminol acted as a solid support for CA125 primary antibody, and GOx–GHS was employed as the supporter of CA 125 secondary antibody. With the addition of glucose, the hydrogen peroxide was prepared and the ECL property of the AuNPs–EGN–luminol was enhanced. The proposed immunosensor enabled CA125 concentrations to be determined in the range of 1 pg mL−1 to 100 ng mL−1, with a detection limit of 0.38 pg mL−1.
1. Introduction
To date, many inspection methods, including fluorescence immunoassay,1,2 radioimmunoassay,3 enzyme-linked immunosorbent assay (ELISA),4 mass spectrometric immunoassay,5,6 electrophoretic immunoassay,7 and chemiluminescence immunoassay, have been used for the detection of cancer antigen 125 (CA125) in clinical serum samples. Among various detection methods, electrochemiluminescence (ECL) that represents a fusion between electrochemical and chemiluminescent methods has become an important and powerful tool in analytical and clinical applications because of its simplicity, low cost, and high sensitivity.8,9 Moreover, ECL has several advantages over other analytical approaches because it does not require a light source, which results in a simple instrumentation and low- or zero-background signal. In view of the aforementioned superior characteristics, ECL has been paid considerable attention during the past several decades.10ECL detection with luminol has attracted much attention in bioanalytical and analytical chemistry.11 In addition, the luminol–hydrogen peroxide system has been well-known to produce strong ECL at positive potentials.12 We have demonstrated that the utilization of cost-effective luminol–hydrogen peroxide system could overcome the disadvantages of conventional ECL signal probes by simplifying analytical procedure and shortening analysis time. In addition, the previous works indicate that the ECL behavior of luminol is strongly dependent on the materials and surface status of the electrodes involved in the ECL experiments. The luminol ECL on a platinum or gold NP-modified electrode has been reported, and the luminol ECL process could be accelerated due to the catalytic property of the Pt or AuNPs.13,14
Recently, gold nanoparticles (AuNPs) have attracted considerable attention as one of the most popular and promising conducting materials because of its large specific surface area, good stability and interesting electrical and optical properties15, and biocompatibility.16,17 More importantly, AuNPs have provided a good pathway of electron transfer, and they have enhanced the immobilized amount of biomolecules.18 In addition, considerable attention has been paid to exfoliated graphene (EGN) due to its fascinating two-dimensional structure, unusual electrochemical properties, and large accessible surface area.19,20 Because of the large surface area of EGN and related derivatives it can also be used as an excellent carrier for loading more active probes for binding biomolecules, offering a significant amplification on the electrochemical sensing signals.21 Herein, the AuNPs are immobilized on the surface of EGN through electrostatic incorporation using poly(diallyldimethylammonium chloride) (PDDA), fabricating the gold nanoparticles–exfoliated graphene (AuNPs–EGN) composites. The hydroxyl of EGN can connect with luminol through N,N′-carbonyl diimidazole (CDI).
Recently, ECL-based detection has played a significant role at the forefront of the bioanalysis area because of its ultrahigh sensitivity and selectivity. Furthermore, the ECL strategy can decrease the detection limit to the single-molecular level. Because enzymes exhibit outstanding specificity, selectivity, and catalytic activity, these merits have led to their widespread applications in research, medicine, and industry.22–24 To date, enzymes have been widely applied as recognition and signaling elements for the detection of some specific molecular analytes.25,26 Glucose oxidase (GOx) can catalyze the oxidation of glucose to gluconolactone along with the generation of hydrogen peroxide (H2O2).27 By quantifying the amount of H2O2 generated, one can indirectly quantify the amount of GOx. Here, we observed that H2O2 can efficiently increase the ECL of the intensity of luminol. Therefore, an ECL immunosensor based on the GOx–GHS system is available.
A signal-type ECL sandwich immunosensor for the detection of CA125 has been prepared. In the immunosensor, the AuNPs–EGN–luminol acted to be the solid support for CA125 primary antibody (Ab1), and CA125 secondary antibody (Ab2) was incubated on the surface of the GOx–GHS nanoparticles. Thus, we obtained the ECL intensities, and thus the amount of CA125 could be ascertained.
2. Experiments
2.1. Reagents
All the chemicals and solvents used were of analytical grade or of the highest purity available. CA125, Ab1 and Ab2, and bovine serum albumin (BSA, 96–99%) were obtained from Shanghai Linc-Bio Science Co. Ltd (Shanghai, China). Luminol, dimethyl sulfoxide (DMSO), CDI, poly(diallyldimethylammonium chloride) (PDDA, 20%, w/w in water, Mw 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–350000) and bis(2-ethylhexyl) sodium sulfosuccinate (AOT) were obtained from Sigma-Aldrich (St. Louis, Mo., USA). HAuCl4·4H2O, hydrazine hydrate (80%), GOx (100–250 units per mg) and sodium citrate were purchased from Shanghai Chemical Reagent Company (Shanghai, China).
000–350000) and bis(2-ethylhexyl) sodium sulfosuccinate (AOT) were obtained from Sigma-Aldrich (St. Louis, Mo., USA). HAuCl4·4H2O, hydrazine hydrate (80%), GOx (100–250 units per mg) and sodium citrate were purchased from Shanghai Chemical Reagent Company (Shanghai, China).
All solutions were prepared using Milli-Q water (Millipore) as a solvent. Phosphate buffered solutions (PBS, pH 7.4) were prepared using 0.01 mol L−1 KH2PO4 and 0.01 mol L−1 Na2HPO4. PBS (0.01 mol L−1, pH 7.4), containing 1% glucose, was used as the electrolyte. The CA125 was stored at 4 °C, and its standard solution was daily prepared with PBS solution when required. The clinical serum samples were provided by Shandong Tumor Hospital.
2.2. Apparatus
The ECL measurements were carried out using a MPI-E multifunctional electrochemical and chemiluminescent analytical system (Xi'an Remex Analytical Instrument Ltd, Co.) with the voltage of the photomultiplier tube (PMT) set at 800 V. Cyclic voltammetric measurements (CVs) were performed with a CHI 760D electrochemical workstation (Shanghai CH Instruments, China). Electrochemical impedance spectroscopy (EIS) was carried out on an IM6x electrochemical station (Zahner, Germany). Transmission electron microscopy (TEM) images were obtained using a Hitachi H-800 microscope (Japan). The morphologies of AuNPs–EGN and GOx–GHS were characterized with scanning electronic microscopy (SEM) on a QUANTA FEG 250 thermal field-emission scanning electron microscope (FEI Co., USA). UV-vis experiments were performed with a UV-3900 spectrophotometer (Hitachi, Japan). X-ray diffraction (XRD) patterns were obtained using a D8 Advance (Bruker) X-ray diffractometer with Cu KR radiation (λ = 1.5418 Å). All the experiments were carried out with a conventional three-electrode system with a modified glassy carbon electrode (GCE, 3 mm in diameter) as the working electrode (WE), a platinum counter electrode (CE) and an Ag/AgCl (sat. KCl) reference electrode (RE).2.3. Preparation of PDDA–EGN
First, a modified Hummers method28,29 was applied to synthesize the graphene oxide (GO) from natural graphite powder. Then, the as-prepared GO was exfoliated by ultrasonication for more than 2 h, and the obtained dispersion was then subjected to 10 min of centrifugation at 10000 r·min− to get a black-brown supernatant (0.5 mg mL−1). Finally, the homogeneously exfoliated GO (EGO) aqueous dispersion was reduced into PDDA–EGN.
In a typical procedure, to a three-necked flask containing 100 mL EGO solution (0.50%), 500 μL PDDA (20%) solution was added and stirred for 30 min. Next, 500 μL hydrazine hydrate (80%) was added and subjected to stirring for 24 h at 90 °C. Finally, excess of PDDA was removed by centrifugation, and the composite was rinsed twice with water, and the black PDDA–EGN was then readily redispersed in water by mild ultrasonication, forming a black suspension with a final concentration of 1.0 mg mL−1.
2.4. Composition of AuNPs–EGN–luminol hybrids
AuNPs with a diameter of ∼13 nm were produced by reducing gold chloride tetrahydrate with citric acid at 96 °C for 15 min. AuNPs should be dispersed by ultrasonication prior to use. In a typical experiment of self-assembly, 100 μL of PDDA–EGN (1.00 mg mL−1) was added into 5 mL of AuNPs solution (0.10 mg mL−1) with stirring and ultrasonic processing for 10 min to form a homogeneous mixture. The mixture was then kept undisturbed under ambient conditions for 12 h. Finally, the precipitate was collected by centrifugation with water for several times. The as-prepared AuNPs–EGN hybrids were redispersed in 1 mL of water, and the final concentration obtained was 0.10 mg mL−1.Preparation of AuNPs–EGN–luminol: first, the AuNPs–EGN hybrids were dried under vacuum at 60 °C for 12 h, and the composite was dipped in 5 mL of CDI solution (1%) at room temperature for 4 h. Subsequently, the precipitate was collected by centrifugation. The mixture was then dispersed into 5 mL of luminol solution (1.00 × 10−4 mol L−1) and kept undisturbed under ambient conditions for 4 h. Finally, the precipitate was collected by centrifugation with water for several times. The as-prepared AuNPs–EGN–luminol was redispersed in 1 mL of distilled water.
2.5. Preparation of Ab1–AuNPs–EGN–luminol
To generate AuNPs–EGN–luminol composite immunological labels, 1 mL of the above AuNPs–EGN–luminol suspension was mixed with 1 mL of Ab1 solution (20 μg mL−1 of Ab1 in 0.01 mol L−1 PBS) (pH 7.4). After incubation for 4 h, the residual antibody was removed by centrifugation and washing with PBS (0.1 mol L−1) for several times. Subsequently, excess of antibodies were washed with PBS (pH 7.4) and incubated in 1% BSA for 1 h to block non-specific binding sites. The resultant Ab1–AuNPs–EGN–luminol composite was dispersed with PBS (pH 7.4) to a final volume of 2 mL and stored at 4 °C for subsequent usage.2.6. Preparation of GOx–GHS-labeled Ab2
First, the preparation of GOx–GHS composite: the GOx–GHS were synthesized according to the literature30 with a little modification. Two solutions were initially prepared as follows: solution A, including 200 μL of 0.05 mol L−1 CaCl2, 100 μL of 1.50 × 10−4 mol L−1 GOx aqueous solution, and 15 mL of 0.10 mol L−1 AOT isooctane suspension, and solution B, containing 200 μL of 0.05 mol L−1 Na2CO3, 100 μL of 1.50 × 10−4 mol L−1 GOx aqueous solution, and 15 mL of 0.10 mol L−1 AOT isooctane suspension. After adequately stirring the GOx, solution B was added dropwise into solution A with violent stirring until the translucent GOx-doped CaCO3 colloids were formed. Following that 100 μL of 5.00% HAuCl4 solution was added dropwise into the reverse micellar solution. After stirring the mixture further for 2 h at room temperature, 100 μL of 0.05 mol L−1 aqueous hydrazine hydrate was injected into the prepared suspension. With the aid of NH2OH, the Au3+ ions coating the CaCO3 surface were reduced to metallic Au0. With the progressive addition of hydrazine hydrate to reverse micelles, the solution acquired a fawn-to-red color due to the formation of gold colloids. After stirring for 1 h at room temperature, 3 mL of absolute ethanol was added and stirred for 10 min, which resulted in the complete breakdown of reverse micelles with the formation of two immiscible layers of aqueous ethanol and isooctane. The ethanol was carefully removed using a separating funnel. The particles thus obtained were washed four times with isooctane and centrifuged to remove any residual AOT. The pelleted particles were then dispersed in 10 mL of water by vigorous stirring for 30 min, and the dispersed system was dialysed against distilled water for 2 h using a 12 kDa cut-off dialysis bag. The hollow microspheres of nanogold particles were finally formed by the addition of 0.001 mol L−1 HCl into the nanogold coated GOx-doped-CaCO3 suspension. Thus, GOx–GHS was prepared.Second, to generate GOx–GHS composite immunological labels, 1 mL of the aforementioned GOx–GHS suspension was mixed with 1 mL of Ab2 solution (20 μg mL−1, pH 7.4). After incubation for 5 h, the residual Ab2 was removed by centrifugation and washed several times with 0.1 mol L−1 PBS. Subsequently, the GOx–GHS composite-labeled Ab2 was redispersed in 2 mL of 1% BSA solution for 2 h to block the excess of amino groups and nonspecific binding sites of the GOx–GHS composite-labeled Ab2. After being centrifuged and washed with PBS, the resultant GOx–GHS composite-labeled Ab2 was dispersed in 0.01 mol L−1 of pH 7.4 PBS to a final volume of 2 mL and stored at 4 °C for subsequent usage.
2.7. Fabrication of the sandwich-type electrochemiluminescence immunosensor
The entire process for constructing the modified electrode is schematically shown in Scheme 1. A GCE electrode with a 3 mm diameter was carefully polished with 1.0, 0.3 and 0.05 mm alumina powder on fine abrasive paper and ultrasonically washed with water. First, 5 μL Ab1–AuNPs–EGN–luminol hybrids were coated on the working electrodes and dried at room temperature for 30 min. The electrode was then incubated with various concentrations of CA125 solution for 30 min. Subsequently, the prepared GOx–GHS composite-labeled Ab2 was dropped onto the electrode surface and incubated for another 30 min. Finally, the electrode was washed with PBS (pH 7.4), and the immunosensor was prepared. | ||
| Scheme 1 Fabrication process of the electrochemical immunosensor. | ||
2.8. ECL detection of CA125 with the immunosensor
ECL detection of CA125 with the immunosensor ECL measurements was done at room temperature. The modified electrode was dipped in the 0.01 mol L−1 PBS containing 1% glucose, and after a reaction of 10 min, the ECL intensity was measured. The potential swept from 0 to 0.6 V with a scan rate of 50 mV s−1 with a photomultiplier tube voltage of −800 V. The ECL signals related to the CA125 concentrations could then be measured.3. Results and discussion
3.1. Characterization of AuNPs–EGN–luminol composite
SEM was used to characterize the prepared EGN (Fig. 1A), which have a large surface area for loading more AuNPs. In addition, the SEM revealed the structure of AuNPs–EGN. As shown in Fig. 1C and D, AuNPs with a diameter of ∼13 nm (Fig. 1B) were dispersed on the surface of EGN, and some of them are covered with EGN. In addition, AuNPs could also be found inside the cavity of EGN, which implied that the AuNPs were dispersed in a three-dimensional structure along with EGN, and no aggregation of EGN sheets or free AuNPs was observed, which suggested that the AuNPs almost absorbed in to the EGN. The unique structure of the prepared composite couple be might benefit the electrochemical or ECL applications. | ||
| Fig. 1 (A) SEM image of EGN, (B) TEM image of AuNPs, (C) SEM image of AuNPs–EGN, (D) magnified SEM image of AuNPs–EGN. | ||
To monitor the formation of AuNPs–EGN–luminol, we studied the UV-vis absorption spectra of the luminol, AuNPs, and AuNPs–EGN–luminol, respectively (Fig. 3A). The pure luminol showed absorption peaks at 301 and 348 nm (curve b), whereas there was a peak at 523 nm for the pure AuNPs (curve a). When luminol and AuNPs were encapsulated into the EGN, three absorption peaks were simultaneously observed (curve c). The peaks at 301 and 349 nm were mainly ascribed to the luminol, whereas the peak at 522 nm was attributed to the AuNPs. Compared with those obtained with pure luminol solution and pure AuNPs, the slight deviation between absorption wave numbers was due to the interaction between AuNPs–EGN and the luminol solution. Thus, we could conclude that the AuNPs–EGN–luminol composite were prepared.
3.2. Characterization of GOx–GHS composite
In this study, the GHS provided a large area for the encapsulation of the inner GOx, and hundreds to thousands of GOx molecules were adsorbed on one GHS. Furthermore, it also provided a larger area for the immobilization of the external Ab2. When appropriately applied in bioanalysis, the GOx–GHS provided a significant improvement in analytical sensitivity. The SEM and magnified SEM images of GOx–GHS are shown in Fig. 2A and B, and the TEM image of GOx–GHS is shown in Fig. 2C, which displayed good dispersion with a mean diameter of ∼50 nm. | ||
| Fig. 2 SEM (A) and magnified SEM (B) image of GOx–GHS, (C) TEM image of GOx–GHS. | ||
In addition, to the binding energy differences, we also observed the geometrical differences between the considered systems, as measured by 2θ. As shown in the XRD of GOx–GHS (Fig. 3B), for the systems of GOx–GHS, the curve exhibited two diffraction peaks at 38.2° and 44.6°, which can be indexed to diffraction from the (111) and (220) of the fcc structure of metallic Au, respectively. To further monitor the formation of GOx–GHS, the UV-vis absorption spectrum of the GOx–GHS was also investigated. The GOx (curve d on Fig. 3A) showed absorption peaks at 377 and 453 nm. When GOx–GHS were prepared (curve e on Fig. 3A), the peaks at 379 and 445 nm were mainly ascribed to the GOx molecules, whereas the peak at 522 nm was attributed to the AuNPs. Thus, we could conclude that GOx molecules and colloidal gold could be doped into the synthesized nanocomposites via the reverse micellar method.
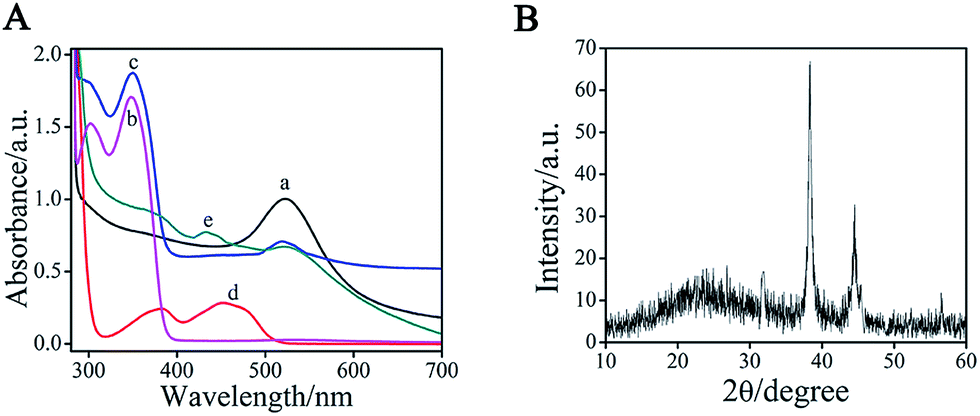 | ||
| Fig. 3 (A) UV-vis absorption spectra of (a) AuNPs, (b) luminol, (c) AuNPs–EGN–luminol composite, (d) GOx, and (e) GOx–GHS composite. (B) XRD of AuNPs-EGN. | ||
3.3. Possible mechanisms for ECL production
The mechanisms for ECL production of the fabricated immunosensor were studied and inferred as follows: first, glucose reduced the flavin adenine dinucleotide (FAD) of GOx to reduced FAD (FADH2), simultaneously forming gluconic acid. Second, FADH2 obtained was reoxidized by oxygen dissolved in the solution, and as a result H2O2 was produced. Third, under the weak alkaline condition, the H2O2 molecules were mostly converted to HOO−. Fourth, the luminol on the surfaces of the EGN–AuNPs nanoparticles were excited by electron (e−), forming the excited state of luminol (luminol*). Fifth, HOO− could transfer the energy of luminol* (a), generating a stable-state luminol. Finally, the remaining luminol* (b) could emit light (hν). The possible mechanism of ECL can be described by the following equations:| GOx (FAD) + 2 glucose → 2 gluconic acid + GOx (FADH2) | (1) |
| GOx (FADH2) + O2 → GOx (FADH2) + H2O2 | (2) |
| H2O2 + OH− → HOO− + H2O | (3) |
| Luminol + e− → luminol* | (4) |
| Luminol* (a) + 2HOO− → luminol + 2OH− + O2 | (5) |
| Luminol* (b) → luminol + hν | (6) |
3.4. ECL and EIS behaviors of the immunosensor
To investigate the technique of the AuNPs–EGN–luminol composites for ECL analysis, we designed two experiments. One experiment was designed without the EGN for the sensing platform, the other without the AuNPs for the signal label. In Fig. 4A, we compared the ECL intensity of AuNPs–luminol composite-labeled Ab1 and AuNPs–EGN–luminol composite-labeled Ab1. As can be seen, AuNPs–EGN–luminol composites could increase the ECL, which could benefit the immobilization analysis. In the Fig. 4B, we compared the ECL intensity of EGN–luminol composite-labeled Ab1 and AuNPs–EGN–luminol composite-labeled Ab1. The AuNPs–EGN–luminol composite-modified GCE reveals excellent ECL performance compared to EGN–luminol composites. | ||
| Fig. 4 (A) The immunosensor performance with (a) and without (b) EGN. (B) The immunosensor performance with (a) and without (b) AuNPs. | ||
To verify the enzymatic amplification of the immunosensor, the ECL behavior of the GCE/Ab1–AuNPs–EGN/CA125/GOx–GHS–Ab2 electrode was studied by adding 1% glucose into the aforementioned PBS solution, and the ECL signal of luminol showed a sharp increase (curve b in Fig. 5A). Compared with the ECL behavior without the addition of the glucose (curve a on Fig. 5A), the intense ECL signal for the as-prepared electrode originated from the synergistic effect of the H2O2, which resulted from the GOx-mediated electrooxidation of glucose in the presence of oxygen. On the whole, the multiple enzymatic turnovers significantly amplified the generated ECL photons.
 | ||
| Fig. 5 (A) GOx–GHS–Ab2/CA125/Ab1/AuNPs–EGN/GC electrode (without (curve a) and with (curve b) 1% glucose) in 0.1 mol L−1 PBS solution (pH 7.4). (B) EIS of bare GCE (a), GCE/Ab1–AuNPs–EGN–luminol (b), GCE/Ab1–AuNPs–EGN–luminol/CA125 (c), GCE/Ab1–AuNPs–EGN–luminol/CA125/GOx–GHS–Ab2 (d) in 0.01 mol L−1 PBS (2.5 mmol L−1 Fe(CN)64−/3− + 0.1 mol L−1 KCl, pH 7.4). The frequency range is between 0.01 and 100000 Hz with signal amplitude of 5 mV. (C) ECL profiles of the immunosensor at various pH of PBS containing 1% glucose. (D) The potential range (V): (a) 0–0.5, (b) 0–0.55, (c) 0–0.6, and (d) 0–0.65. The CA125 concentration was 5 ng mL−1. Scan rate: 100 mV s−1. The voltage of the photomultiplier tube was set at 800 V. | ||
EIS is an effective method to monitor the changes of interfacial properties, which allow understanding the chemical transformation and processes associated with the conductive electrode surface.31–33 In EIS, the diameter of the semicircle at higher frequencies corresponds to the electron-transfer resistance (Ret); a change in the value of Ret was associated with the blocking behavior of the modification processes in the GCE, which was reflected in the EIS as a change in the diameter of the semicircle at high frequencies. The EIS of the electrodes were performed in a background solution of 5.0 mmol L−1 K3Fe(CN)6 containing 0.1 mol L−1 KCl, and the frequency range was at 100 mHz to 10 kHz with a signal amplitude of 5 mV. Fig. 5B shows the EIS of the GCE at different stages. The EIS of bare GCE revealed a relatively small semicircle domain (curve a). When Ab1–AuNPs–EGN–luminol was modified on the GCE surface, a much lower resistance was obtained (curve b), implying that the AuNPs–EGN–luminol is an excellent electrical conducting material and accelerates the electron transfer. Furthermore, CA125 could resist all the electron-transfer kinetics of the redox probe at the fiber surface, resulting in the increase of resistance (curve c), which verified the immobilization of these substances. Finally, when GOx–GHS–Ab2 interacted with CA125 (curve d), the resistance decreased, indicating that the synthesized GOx–GHS possessed high conductivity and good electron transfer efficiency, although the protein adsorption layer acted as a barrier to the interfacial electron transfer.
3.5. Optimization of experimental conditions
To achieve an optimal ECL signal, the pH value of the substrate solution was an important factor for the ECL intensity. According to the experimental results in Fig. 5C, the ECL intensity of the immunosensor is obviously affected at different pH, which may be a result of the chemical properties of the immunosensor varying with the changes of acidity. Obviously, PBS (0.01 mol L−1, pH 7.8) can be selected as the detection solution. Fig. 5D shows that the ECL intensity is responsive to different potentials (outlined in Fig. 5D). It can be observed that the optimal scanning potential is 0–0.6 V.3.6. Analytical performance
Under the optimal conditions, the proposed immunosensor was applied to detect the different concentrations of CA125. As shown in Fig. 6B and C, the ECL intensity of the immunosensor increased with the increasing CA125 concentrations, and it exhibited a good linear relationship with the logarithm of CA125 concentration from 0.001 to 100 ng mL−1. The linear regression equation can be given as IECL = 1345.62 + 410.63logcCA125 (cCA125/ng mL−1), with a correlation coefficient of 0.9921. The detection limit value for CA125 was determined at 0.38 pg mL−1, estimated at a signal-to-noise ratio of three criterion, which suggested a satisfactory detection limit and linear range.
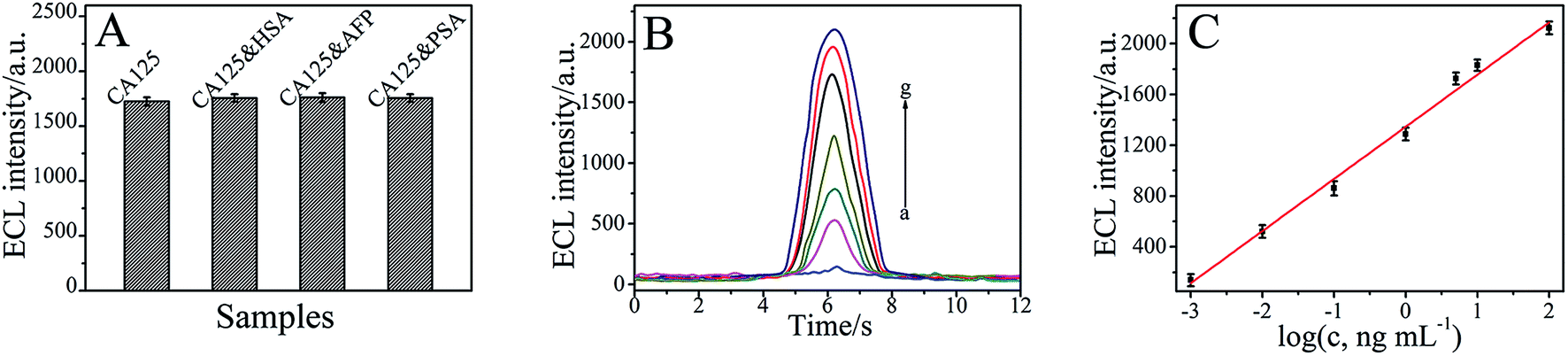 | ||
| Fig. 6 (A) Specificity of the immunosensor. (B) ECL profiles of the immunosensor in the absence (a–g) of different concentrations of CA125 in PBS solution containing 1% glucose at pH of 7.4. CA125 concentration (ng mL−1): (a) 0.001, (b) 0.01, (c) 0.1, (d) 1, (e) 5, (f) 10, and (g) 100 (the voltage of the photomultiplier tube was set at 800 V). (C) The calibration curve of the immunosensor. | ||
Table 1 shows the linear range and detection limit of immunosensors along with those of previous reports.34–37 Compared with other methods,38–40 the as-prepared immunosensor has a relatively large linear range and low detection limit. The results demonstrated that the proposed method has huge application potential in the field of cancer diagnosis.
3.7. Specificity, stability, and reproducibility of the immunosensor
To further characterize the specificity of the immunosensor, 5 ng mL−1 CA125, containing 200 ng mL−1 human serum albumin (HSA), 200 ng mL−1 alpha fetoprotein (AFP), and 200 ng mL−1 prostate protein antigen (PSA) was, analysed by the immunosensor, and the results are shown in Fig. 6A. The relative deviations (RD) were 1.86%, 2.08% and 1.79%. These results indicated that the relative standard deviations are all less than 3.00%, which suggested that the specificity of the as-prepared immunosensor was acceptable.The stability of this immunosensor was tested by carrying out 10 continuous cyclic scans in 0.1 mol L−1 PBS (pH 7.4). In addition, when the immunosensor was dried and stored at 4 °C, the ECL response retained 94.12% of the initial response after a storage period of 30 days. The slow decrease in response may be related to the gradual deactivation of the immobilized antibody incorporated in the composite.
The reproducibility played a vital role in the performance of the prepared immunosensor. When the ECL response for CA125 of six fabricated immunosensors was tested, the result showed a relative standard deviation of 3.12%, giving an acceptable fabrication reproducibility of the immunosensors.
3.8. Application of ECL immunosensor in human serum samples
The feasibility of the immunoassay system in clinical applications was investigated by analyzing several real samples. In comparison with the ELISA method, these serum samples were diluted to various concentrations with PBS of pH 7.4.Table 2 describes the correlation between the results obtained by the proposed ECL immunosensor and the ELISA method. It clearly indicates that there is no significant difference between the results and the ELISA method. Thus, the developed immunosensor could be satisfactorily applied to the clinical determination of CA125 levels in human serum.
| Immunosensor concentrations (ng mL−1) | ELISA concentrations (ng mL−1) | ||||||
|---|---|---|---|---|---|---|---|
| Sample | Added | Found | Recoveries (%) | Added | Found | Recoveries (%) | Relative deviation (%) |
| 1 | 0.5 | 0.507 | 101.4 | 0.5 | 0.512 | 102.4 | −0.99 |
| 2 | 1.0 | 0.983 | 98.3 | 1.0 | 0.976 | 97.6 | 0.712 |
| 3 | 2.0 | 2.022 | 101.1 | 2.0 | 2.036 | 101.8 | 0.69 |
| 4 | 3.0 | 3.044 | 101.5 | 3.0 | 3.031 | 101.0 | −0.49 |
4. Conclusions
This article describes an ECL immunoassay for CA125 measurement. The AuNPs–EGN–luminol with uniform pore size, large surface area and good biocompatibility can be simply prepared, with the GOx–GHS composite as an ideal label, which has excellent labeling properties and ECL activity with amplification techniques. Although the present assay system is focused on the determination of the target antigen molecules, it can be easily extended to the detection of other antigens or biocompounds. Moreover, the potential of this method for application is its simple and efficient diagnostic strategy for immunoassays. Importantly, this approach does not require sophisticated fabrication, and it is well suited for high-throughput biomedical sensing and application in both clinical and biodefense areas.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (51273084, 21277058), Natural Science Foundation of Shandong Province, China (ZR2012BZ002), and Technology Development Plan of Shandong Province, China (Grant no. 2012GGB011813).References
- T. Kreisig, R. Hoffmann and T. Zuchner, Anal. Chem., 2011, 83, 4281–4287 CrossRef CAS PubMed.
- T. Ruckstuhl, C. M. Winterflood and S. Seeger, Anal. Chem., 2011, 83, 2345–2350 CrossRef CAS PubMed.
- T. D. Eller, D. R. Knapp and T. Walle, Anal. Chem., 1983, 55, 1572–1575 CrossRef CAS.
- V. Jimenez, J. Adrian, J. Guiteras, M. P. Marco and R. Companyo, J. Agric. Food Chem., 2010, 58, 7526–7531 CrossRef CAS PubMed.
- O. Trenchevska, E. Kamcheva and D. Nedelkov, J. Proteome Res., 2010, 9, 5969–5973 CrossRef CAS PubMed.
- S. L. Hu, S. C. Zhang, Z. C. Hu, Z. Xing and X. R. Zhang, Anal. Chem., 2007, 79, 923–929 CrossRef CAS PubMed.
- C. L. Hou and A. E. Herr, Anal. Chem., 2010, 82, 3343–3351 CrossRef CAS PubMed.
- Y. Zhang, S. G. Ge, S. W. Wang, M. Yan, J. H. Yu, X. R. Song and W. Y. Liu, Analyst, 2012, 137, 2176–2182 RSC.
- Y. Zhang, W. Y. Liu, S. G. Ge, M. Yan, S. W. Wang, J. H. Yu, N. Q. Li and X. R. Song, Biosens. Bioelectron., 2013, 41, 684–690 CrossRef CAS PubMed.
- C. S. Pinaud, F. Beutel, O. You, C. Piehler and J. Dahan, Nano Lett., 2010, 10, 2147–2154 CrossRef PubMed.
- S. Xu, Y. Liu, T. H. Wang and J. H. Li, Anal. Chem., 2011, 83, 3817–3823 CrossRef CAS PubMed.
- H. Cui, W. Wang, C. F. Duan, Y. P. Dong and J. Z. Guo, Chem.–Eur. J., 2007, 13, 6975–6984 CrossRef CAS PubMed.
- X. Chen, Z. Lin, Z. Cai, X. Chen, M. Oyama and X. J. Wang, Nat. Nanotechnol., 2009, 9, 2413–2420 CAS.
- X. Liu, W. Niu, H. Li, S. Han, L. Hu and G. Xu, Electrochem. Commun., 2008, 10, 1250–1253 CrossRef CAS PubMed.
- G. S. Lai, J. Wu, H. X. Ju and F. Yan, Adv. Funct. Mater., 2011, 21, 2938–2943 CrossRef CAS.
- S. G. Xu, H. Yuan, A. Xu, J. Wang and L. J. Wu, Langmuir, 2011, 27, 13629–13634 CrossRef CAS PubMed.
- Y. He and H. Cui, J. Phys. Chem. B, 2012, 116, 12953–12957 CrossRef CAS PubMed.
- J. Zhang, H. G. Nie, Z. Wu, Z. Yang, L. J. Zhang, X. G. Xu and S. M. Huang, Anal. Chem., 2014, 86, 1178–1185 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- Z. Liu, Q. Liu, Y. Huang, Y. Ma, S. Yin, X. Zhang, W. Sun and Y. Chen, Adv. Mater., 2008, 20, 3924–3930 CrossRef CAS.
- D. Du, L. Wang, Y. Shao, J. Wang, M. H. Engelhard and Y. Lin, Anal. Chem., 2011, 83, 746–752 CrossRef CAS PubMed.
- E. Katz, J. Wang, M. Privman and J. Halamek, Anal. Chem., 2012, 84, 5463–5469 CrossRef CAS PubMed.
- Z. Liu, B. Cho, T. Ouyang and B. Feldman, Anal. Chem., 2012, 84, 3403–3409 CrossRef CAS PubMed.
- S. P. Rafael, A. Vallee-Belisle, E. Fabregas, K. Plaxco, G. Palleschi and F. Ricci, Anal. Chem., 2012, 84, 1076–1082 CrossRef CAS PubMed.
- E. Bakker, Anal. Chem., 2004, 76, 3285–3598 CrossRef CAS PubMed.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- Y. Hitomi, T. Takeyasu, T. Funabiki and M. Kodera, Anal. Chem., 2011, 83, 9213–9216 CrossRef CAS PubMed.
- Y. Wang, Y. Li, L. Tang, J. Lu and J. H. Li, Anal. Chem., 2009, 81, 9710–9715 CrossRef CAS PubMed.
- Y. Zhang, M. Su, L. Ge, S. G. Ge, J. H. Yu and X. R. Song, Carbon, 2013, 57, 22–33 CrossRef CAS PubMed.
- R. Kumar, A. Maitra, P. Patanjali and R. Sharma, Biomaterials, 2005, 26, 6743–6753 CrossRef CAS PubMed.
- S. M. Park and J. S. Yoo, Anal. Chem., 2003, 75, 455A–461A CAS.
- H. Chen, J. H. Jiang, Y. Huang, T. Deng, J. S. Li, G. L. Shen and R. Q. Yu, Sens. Actuators, B, 2006, 117, 211–218 CrossRef CAS PubMed.
- M. Prodromidis, Electrochim. Acta, 2010, 55, 4227–4233 CrossRef CAS PubMed.
- D. P. Tang, B. L. Su, J. Tang, J. J. Ren and G. N. Chen, Anal. Chem., 2010, 82, 1527–1534 CrossRef CAS PubMed.
- N. Arakawa, E. Miyagi, A. Nomura, E. Morita, Y. Ino, N. Ohtake, Y. Miyagi, F. Hirahara and H. Hirano, J. Proteome Res., 2013, 12, 4340–4350 CrossRef CAS PubMed.
- Z. Fu, Z. Yang, J. Tang, H. Liu, F. Yan and H. Ju, Anal. Chem., 2007, 79, 7376–7382 CrossRef CAS PubMed.
- J. Wu, Y. Yan, F. Yan and H. Ju, Anal. Chem., 2008, 80, 6072–6077 CrossRef CAS PubMed.
- C. B. Andrew, D. Frank and S. P. J. Higson, Anal. Chem., 2008, 80, 9411–9416 CrossRef PubMed.
- Q. L. Lang, F. Wang, L. Yin, M. J. Liu, V. A. Petrenko and A. H. Liu, Anal. Chem., 2014, 86, 2767–2774 CrossRef CAS PubMed.
- Y. F. Wu, P. Xue, Y. J. Kang and K. M. Hui, Anal. Chem., 2013, 85, 8661–8668 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |