
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceYonemitsu-type condensations catalysed by proline and Eu(OTf)3†
Andrea Renzetti*a,
Emanuela Boffab,
Marco Colazzob,
Stéphane Gérardc,
Janos Sapic,
Tak-Hang Chand,
Hiroshi Nakazawaa,
Claudio Villanie and
Antonella Fontanab
aDepartment of Chemistry, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, 558-8585 Osaka, Japan. E-mail: renzetti@sci.osaka-cu.ac.jp; Fax: +81-6-6605-2522; Tel: +81-6-6605-2513
bDipartimento di Farmacia, Università “G. d'Annunzio” di Chieti-Pescara, Via dei Vestini 33, I-66100 Chieti, Italy
cInstitut de Chimie Moléculaire de Reims, UMR CNRS 6229, Université de Reims Champagne-Ardenne, Faculté de Pharmacie, 51 Rue Cognacq-Jay, F-51096 Reims Cedex, France
dDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, H3A 0B8 Montreal, QC, Canada
eDipartimento di Chimica e Tecnologie del Farmaco, Università “La Sapienza”, P.le A. Moro, I-00185 Rome, Italy
First published on 1st October 2014
Abstract
We report a Yonemitsu-type trimolecular condensation of aromatic heterocycles, aldehydes, and active methylene compounds to afford polyfunctionalised heterocycles. The reaction is catalysed by L-proline and Eu(OTf)3, takes place in methanol at room temperature, and in some cases is highly diastereoselective (d.e. >90%). The reaction offers two advantages with respect to the previously reported Ti(IV)-promoted condensation: (1) it adheres to some principles of green chemistry, and (2) it provides access to compounds that cannot be obtained by classical methodology.
1 Introduction
Green chemistry is the use of safe chemicals and of processes that reduce or eliminate the generation of waste.1,2 In agreement with the twelve principles of green chemistry,2,3 the ideal reaction has 100% yield, is one-pot and catalytic, takes place at room temperature and 1 atm pressure in the air, uses safe and renewable reagents, and generates no waste. Although it is hard to develop a reaction that satisfies all these criteria,4,5 it is possible to make a reaction cleaner. Some years ago, we reported a Yonemitsu-type trimolecular condensation of aromatic heterocycles, aldehydes, and active methylene compounds promoted by Ti(IV) derivatives and Et3N.6,7 The reaction affords polyfunctionalised aromatic heterocycles in a simple one-pot procedure, and can be used to synthesize libraries of heterocyclic compounds in drug discovery.8 However, the Ti(IV)-promoted reaction has two serious drawbacks. Firstly, it uses dangerous reagents: TiCl4 or TiCl2(Oi-Pr)2 (corrosive), Et3N (toxic and flammable), and dichloromethane (carcinogenic). Secondly, it is not catalytic, because it requires equimolar amounts of Ti(IV) salt and Et3N. These compounds are converted into TiOCl2, HCl, and Et3NH+Cl− during the reaction, generating large (stoichiometric) amounts of waste. Due to such limitations, the Ti(IV)-promoted condensation is far from being green, despite being a one-pot process. In this work, we developed a green version of such reaction.2 Results and discussion
2.1 Catalytic system
The first issue we addressed in our study was the development of a catalytic system. To achieve this aim, we used our knowledge on the mechanism of the Ti(IV)-promoted reaction, which involves three steps:9 (1) deprotonation of active methylene compound, (2) Knoevenagel condensation of enolate ion and aldehyde, and (3) Michael addition of heterocycle to the Knoevenagel adduct. The generation of the enolate ion from active methylene compound, being the first step of mechanism, is crucial for the outcome of the overall reaction. Thus, any system that favours the formation of the enolate ion in principle may catalyse the trimolecular condensation.
|
|||
|---|---|---|---|
| Entry | Base | Salt | Yielda (%) |
| a Yield of purified product.b Solvent was CH3CN.c DMAN = 1,8-bis-(N,N-dimethylamino)naphthalene (proton sponge®).d 3,3′-(2-Methylpropane-1,1-diyl)bis(1H-indole) and methyl acetoacetate were recovered.e A complex mixture of compounds was obtained. | |||
| 1 | None | None | 16 |
| 2 | L-Proline | None | 16 |
| 3 | Et3N | None | Traces |
| 4b | DMANc | None | 0d |
| 5 | None | Bi(OTf)3 | 0e |
| 6 | Et3N | Bi(OTf)3 | 38 |
| 7 | DMANc | Bi(OTf)3 | 38 |
| 8 | L-Proline | Bi(OTf)3 | 50 |
| 9 | L-Proline | La(NO3)3·6H2O | 60 |
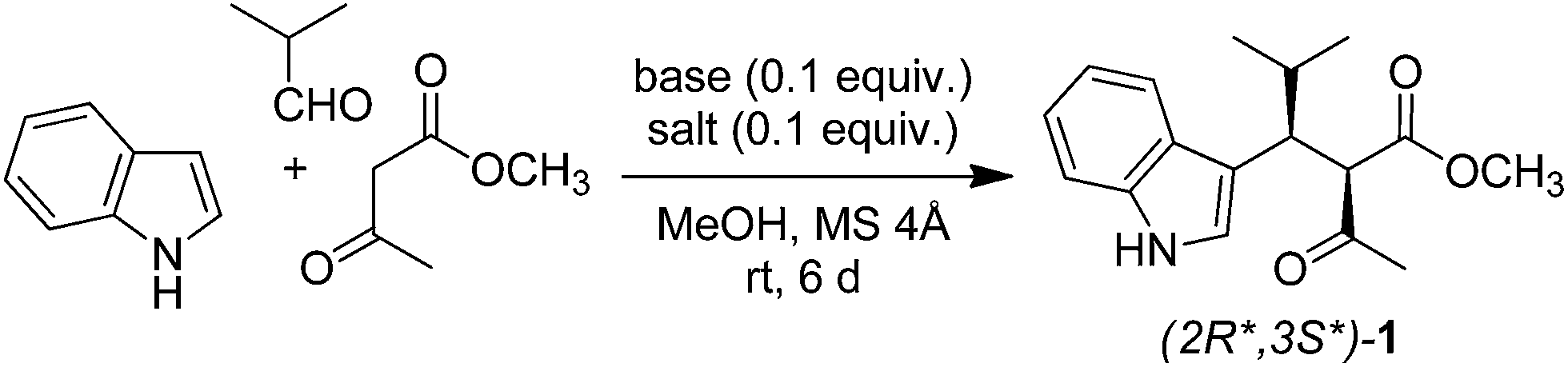
Lewis acids are known to increase the acidity of carbonyl compounds by complexation,23 favouring the generation of enolate ion. Therefore, we studied their use as potential catalysts in our reaction. The first choice fell on bismuth triflate because this salt catalyses Mannich-type reactions,24,25 which are analogous to Yonemitsu-type condensations. Bi(OTf)3 underwent solvolysis26 when used alone, providing a highly acidic (pH = 1) solution and a complex mixture of compounds (Table 1, entry 5). However, the same salt afforded product 1 in 38–50% when used in combination with a Brønsted base (Table 1, entries 6–8). Most importantly, the combination of bismuth triflate and base proved to be more effective than the two compounds used separately. Such synergic effect is likely due to the fact that base stabilizes bismuth by complexation, thus preventing solvolysis and allowing the metal ion to exert its catalytic activity.27 From the above results, it appears that both a Brønsted base and a Lewis acid are necessary to effectively catalyse the reaction. L-Proline proved to be the most efficient base in combination with Bi(OTf)3, affording product 1 in 50% yield (Table 1, entry 8). In fact, L-proline has already been used by Yonemitsu as an efficient catalyst in the three-component condensation of indole, aldehydes, and Meldrum's acid.28 Slightly higher (60%) yield was obtained when Bi(OTf)3 was replaced with La(NO3)3·6H2O (Table 1, entry 9). Solvent screening confirmed methanol as the best reaction medium in the presence of molecular sieves (Table S1†).29
Then, we optimised metal salt (Table 2). We tested several cations maintaining triflate as counter anion owing to its weakly coordinating properties. The highest catalytic activity was displayed by In3+ (Table 2, entry 4) and lanthanide ions, particularly those in the middle of the f-block (Eu3+, Gd3+, and Tb3+; Table 2, entries 13, 14, and 15, respectively). These findings are in line with the ability of such ions to catalyse other multicomponent reactions.30–33 In the case of Tb3+ and Ho3+, the 1H NMR spectrum of the condensation product showed broad signals, indicating complexation with metal (Table 2, entries 15 and 16, respectively).34,35 After filtration over silica gel, the 1H NMR spectrum of 1 displayed sharp signals, confirming the removal of metal. Although terbium triflate gave the best yield (Table 2, entry 15), we kept europium as the elected cation in the following experiments because this ion did not seem to complexate the product according to 1H NMR spectrum.
|
||
|---|---|---|
| Entry | Salt | Yielda (%) |
| a Yield of product after crystallisation.b Conditions: Yb(OTf)3·H2O (0.1 equiv.), neat, sonication, rt, 12 h.c Conditions: L-proline (0.1 equiv.), Eu(OTf)3 (0.1 equiv.), neat, sonication, rt, 12 h. | ||
| 1 | None | 16 |
| 2 | Bi(OTf)3 | 50 |
| 3 | Ga(OTf)3 | 41 |
| 4 | In(OTf)3 | 78 |
| 5 | Mg(OTf)2 | 37 |
| 6 | Sc(OTf)3 | 66 |
| 7 | Y(OTf)3 | 64 |
| 8 | Cu(OTf)2 | 40 |
| 9 | Zn(OTf)2 | 56 |
| 10 | Pr(OTf)3 | 38 |
| 11 | Nd(OTf)3 | 65 |
| 12 | Sm(OTf)3 | 69 |
| 13 | Eu(OTf)3 | 78 |
| 14 | Gd(OTf)3 | 74 |
| 15 | Tb(OTf)3 | 88 |
| 16 | Ho(OTf)3 | 58 |
| 17 | Yb(OTf)3 | 32 |
| 18b | Yb(OTf)3·H2O | Traces |
| 19c | Eu(OTf)3 | 40 |
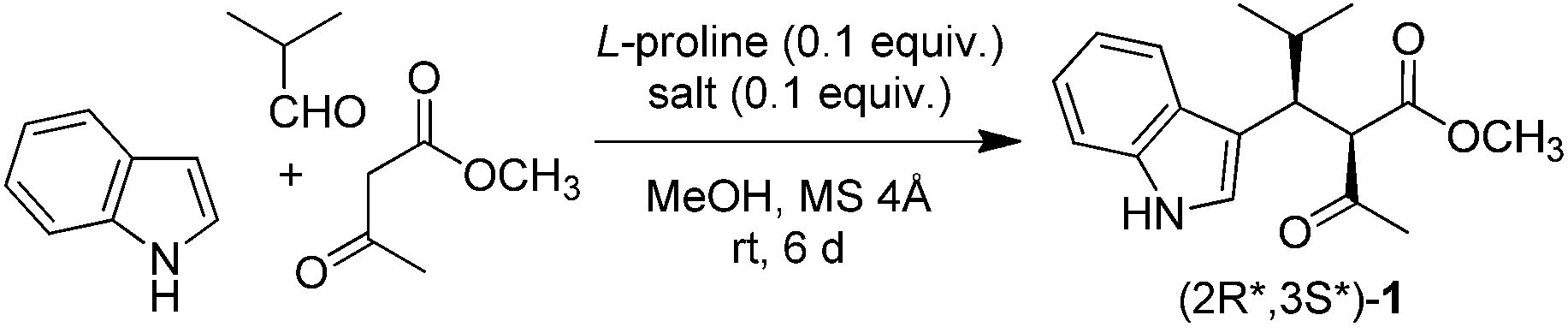
Anion was varied keeping the europium cation constant (Table S2†). The weakly coordinating triflate was found to be the best one. Thus, we selected europium triflate as the metal catalyst in the following experiments. Metal to ligand ratio was optimized as well (Table S3†). The best yield was obtained by using an equimolar ratio of Eu(OTf)3 and L-proline (0.1 equiv. each).
During our work,36 Curini and colleagues reported a trimolecular condensation of dimethyl malonate, indole, and aldehydes catalysed by Yb(OTf)3 monohydrate in neat under ultrasounds.37 Because their reaction is carried out without solvent and base, we checked whether their conditions afford better results than our protocol. The reaction of indole, isobutyraldehyde, and methyl acetoacetate, performed exactly under the same experimental conditions (1.0 mmol scale, 0.1 equiv. of Yb(OTf)3·H2O, neat, ultrasounds, rt, 12 h, same work up) provided 1 only in traces (Table 2, entry 18). We also investigated the effect of sonication in the presence of Eu(OTf)3 (Table 2, entry 19) and L-proline (neat or in MeOH), but in all cases yield was lower than that obtained under our optimised conditions.
2.2 Amino acid
Although L-proline proved to be effective in combination with a Lewis acid, we extended our investigation to other amino acids. The trimolecular condensation of indole, isobutyraldehyde, and methyl acetoacetate was performed in the presence of europium triflate and various L-amino acids as catalysts (Table 3). Some amino acids enhanced the catalytic activity of Eu(OTf)3 (Table 3, entry 1 vs. entries 2, 3, 5, and 8), others inhibited it (Table 3, entry 1 vs. entries 4, 6, and 7). Overall, yield varied from 14 to 78%, indicating that amino acid plays a crucial role in the catalytic activity. However, no correlation between yield and amino acid type was observed. trans-L-4-Hydroxyproline, L-aspartic acid, and L-histidine, bearing three different types of side chain (polar, acidic, and basic, respectively) displayed comparable performance (64, 69, and 59% yield; Table 3, entries 3, 5, and 8 respectively). On the contrary, L-lysine displayed a four-fold lower activity than L-histidine, despite both amino acids possessing a basic side chain (14 and 59% yield; Table 3, entries 6 and 8, respectively). The same result was observed for trans-L-4-hydroxyproline and L-serine, both bearing a hydroxyl group (64 and 20% yield; Table 3, entries 3 and 7, respectively). Proline was the best among the tested amino acids, affording 1 in 78% yield (Table 3, entry 2). When an equimolar mixture of (1S)-(+)-10-camphorsulfonic acid (CSA) and (S)-(+)-1-(2-pyrrolidinylmethyl)pyrrolidine (PMP) was used in the place of an amino acid, 1 was still obtained in 60% yield (Table 3, entry 9). This result indicates that amphiprotic compounds other than amino acids also catalyse the reaction.
|
||
|---|---|---|
| Entry | Amino acid | Yielda (%) |
| a Yield of purified product.b CSA = (1S)-(+)-10-camphorsulfonic acid.c PMP = (S)-(+)-1-(2-pyrrolidinylmethyl)pyrrolidine. | ||
| 1 | None | 50 |
| 2 | L-Proline | 78 |
| 3 | trans-L-4-Hydroxyproline | 64 |
| 4 | L-4-Thiazolidinecarboxylic acid | 29 |
| 5 | L-Aspartic acid | 69 |
| 6 | L-Lysine | 14 |
| 7 | L-Serine | 20 |
| 8 | L-Histidine | 59 |
| 9 | CSAb (0.1 equiv.) + PMPc (0.1 equiv.) | 60 |
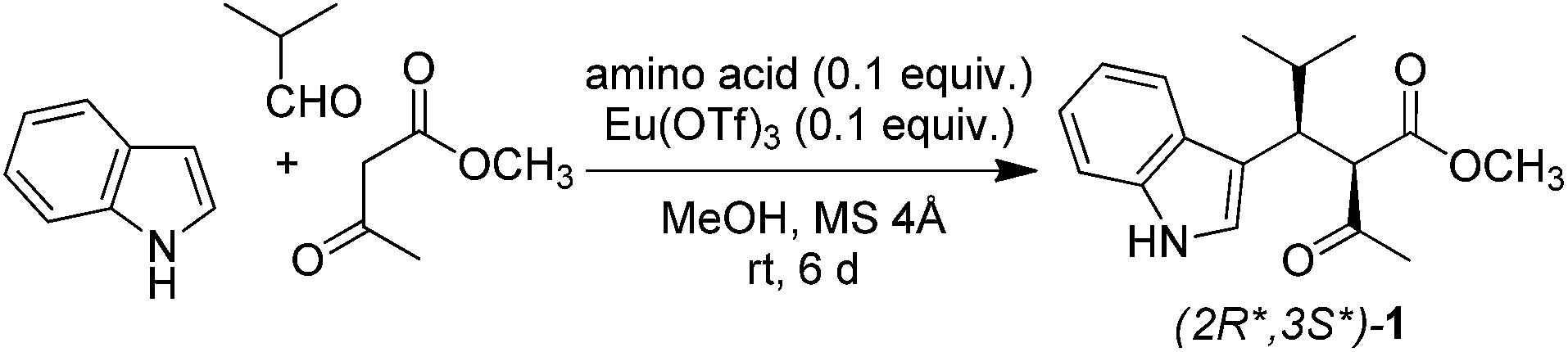
Four representative samples of 1 (entries 2, 5, 8, and 9 of Table 3) were analysed by HPLC on a chiral stationary phase.38 Unfortunately, in all cases the analysis evidenced a racemic mixture, indicating the absence of enantioselectivity.
2.3 Scope and limitations
To check the scope and limitations of our new protocol, we varied the three reactants in turn. Ethyl nitroacetate, ethyl methylsulfonylacetate, and ethyl benzoylacetate as the active methylene compounds were used in ethanol in order to avoid transesterification. Nine condensation products were synthesised (Table 4). All of them were exclusively obtained in the keto form; neither enol nor other tautomeric forms were detected by 1H NMR spectroscopy. This result is in agreement with the well-established evidence that for simple ketones the keto tautomer is favoured in polar solvents such as ethanol.39,40 The absence of any tautomers of the keto form in the synthesised products, in addition to the high solvent polarity, can be attributed to a double effect of the substituent on the α-carbon:41 (1) stabilisation of the keto form by hyperconjugation (electronic effect), and (2) destabilisation of enol as well as other tautomers by repulsion among substituents lying on the same plane (steric effect).
|
|||
|---|---|---|---|
| Entry | Product | Methoda | Yieldb (%) |
| a Method A: L-proline (0.1 equiv.), Eu(OTf)3 (0.1 equiv.), MeOH, MS 4 Å, rt, 6 days. Method B: L-proline (0.1 equiv.), neat, US, rt, 12 h.b Yield of purified product.c Relative stereochemistry has previously been determined.6d Obtained as a single diastereomer; relative stereochemistry was not determined.e Solvent was ethanol.f 50/50 mixture of diastereomers.g 55/45 mixture of diastereomers. | |||
| 1 | 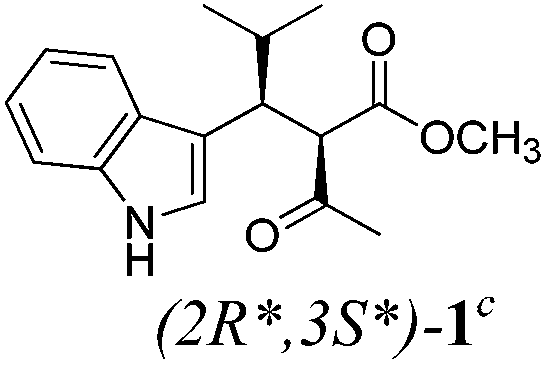 |
A | 78 |
| 2 | 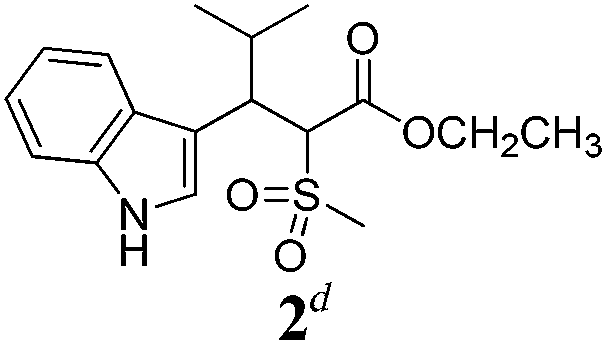 |
Ae | 40 |
| 3 | 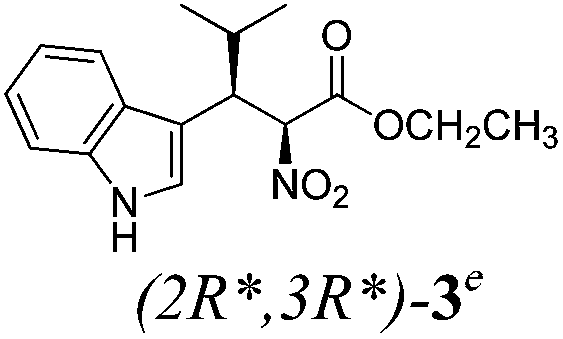 |
B | 38 |
| 4 | 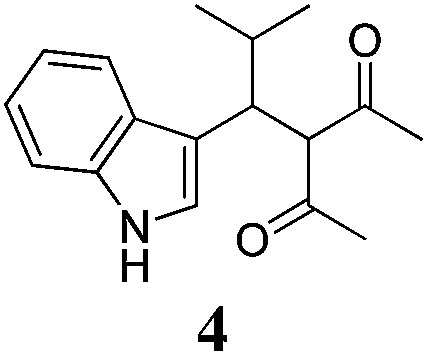 |
A | 15 |
| 5 | 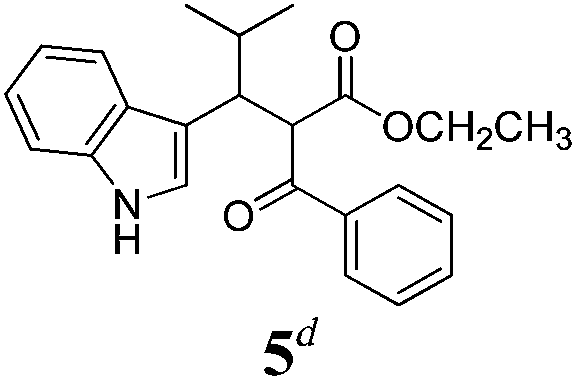 |
Ae | 20 |
| 6 | 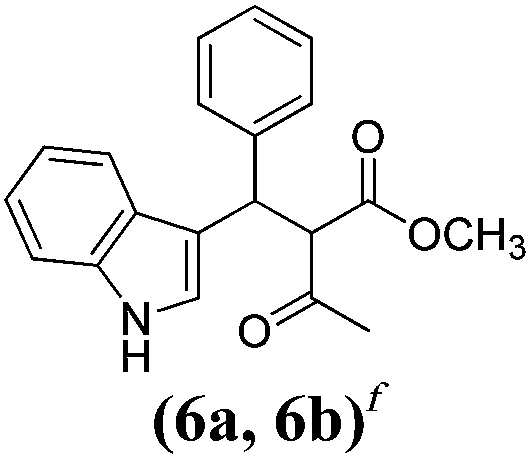 |
A | 26 |
| 7 | 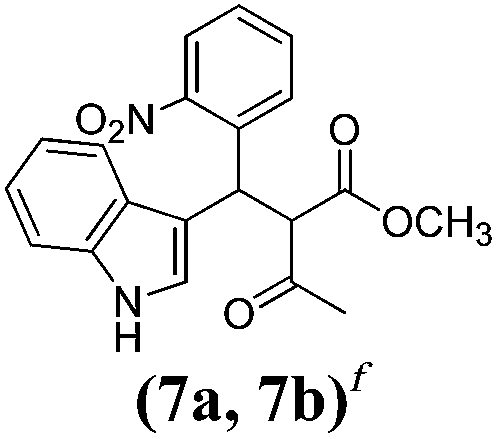 |
A | 35 |
| 8 | 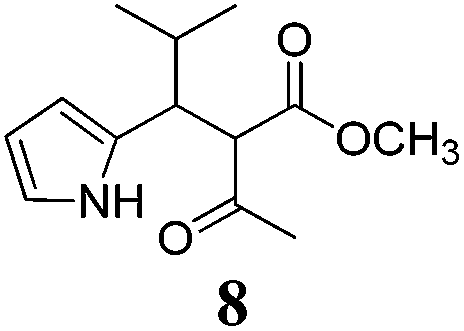 |
A | 47 |
| 9 | 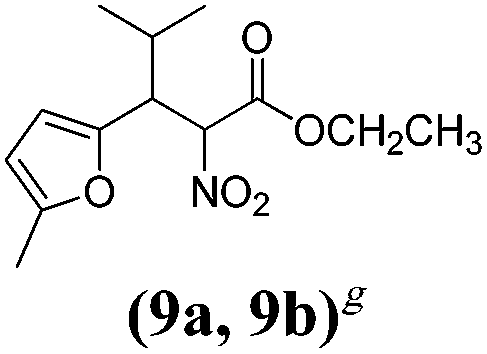 |
A | 35 |
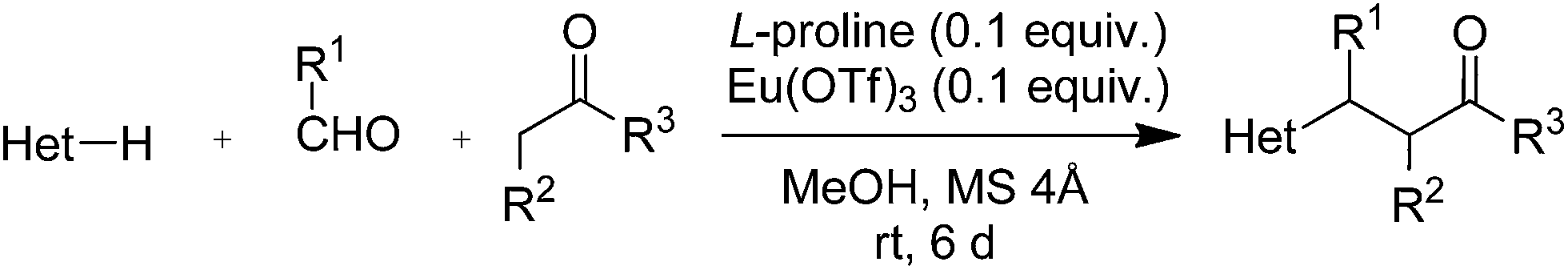
Unfortunately reaction yield was modest in most cases, ranging from 15 to 78%. Ethyl methylsulfonylacetate, acetylacetone, and ethyl benzoylacetate displayed low reactivity despite being stronger acids than methyl acetoacetate, and were recovered largely unreacted (Table 4, entries 2, 4, and 5). Probably their enolate ion was too stabilised by metal ion to undergo the Knoevenagel condensation. Ethyl nitroacetate afforded a mixture of unidentified compounds in the presence of Eu(OTf)3, but reacted in 38% yield when proline was used as the only catalyst (Table 4, entry 3). Under these conditions, ethyl nitroacetate proved to be more reactive than methyl acetoacetate (compare entry 3 in Table 4 with entry 1 in Table 1), probably due to its stronger acidity (pKa = 5.7 and 10.7, respectively). Despite average modest yield, the reaction afforded some compounds (2, 4, 5, and 8) that cannot be obtained by our previously described method based on Ti(IV).6,7,42 The above results illustrate the synthetic utility of this modified Yonemitsu reaction. An alternative method to the synthesis of 3-alkylated indoles is the Michael-type reaction of indole and an α,β-unsaturated carbonyl compound.43–48 This reaction can be performed in high enantiomeric excess using Cu(OTf)2 or Sc(OTf)3 in the presence of a chiral Pybox-type ligand.38,49–51 However, it requires the preliminary isolation and purification of the α,β-unsaturated compound. Our reaction, although not enantioselective, can be performed in one pot, avoiding the isolation of intermediate.
2.4 Mechanism
Two mechanisms can be proposed for the proline/Eu(III)-catalysed trimolecular reaction. The first one (Scheme 1) involves the Knoevenagel condensation of methyl acetoacetate and aldehyde, followed by the Michael addition of indole. In such a pathway, aldehyde and L-proline react to form a highly electrophilic iminium ion (12). Methyl acetoacetate may be either deprotonated by L-proline affording the enolate ion 10 or activated as a β-enaminoester (11) by condensation with L-proline. It is worth noting that deprotonation reaction is an equilibrium because methyl acetoacetate and proline's conjugate acid have almost the same acidity (pKa = 10.7 and 10.6, respectively). The iminium ion 12 is attacked by 10 or 11, and the resulting species 13 eliminates proline to generate the Knoevenagel adduct 14. Eu(III) ion complexates 14, thus favouring the Michael addition of indole to afford the condensation product 1. According to the mechanism proposed in Scheme 1, catalysis results from the combined effect of L-proline and europium: L-proline catalyses the Knoevenagel condensation by activating both nucleophile (β-ketoester) and electrophile (aldehyde); europium catalyses Michael addition by increasing the electrophilicity of the Knoevenagel adduct by complexation. The second mechanism (Scheme 2) involves the Michael addition of the enol of methyl acetoacetate to an azafulvenium ion (18) obtained by the condensation of aldehyde and indole. In this path, Eu(III) ion complexates isobutyraldehyde, thus making aldehyde more electrophilic. Activated aldehyde 16 is attacked by indole generating carbinol 17, which is unstable and rapidly loses water to afford the azafulvenium ion 18. Nucleophilic addition of the enol 20 of methyl acetoacetate to 18 provides the condensation product 1. In the mechanism proposed in Scheme 2, the catalytic activity is attributed only to the metal ion. Both mechanisms are in agreement with some experimental evidences. Knoevenagel adduct 14 was recovered from the crude reaction mixture, and was isolated in 37% yield when methyl acetoacetate and isobutyraldehyde were stirred in the presence of L-proline during 24 h. Reaction of purified 14 with indole in the presence of Eu(OTf)3 afforded the condensation product 1 in 10% yield, confirming that 14 is an intermediate in the trimolecular condensation. The structure of other intermediates can be proposed based on mechanistic studies of analogous reactions. The formation of iminium ion 12 is consistent with the generally accepted mechanism for the Knoevenagel condensation catalysed by secondary bases.52,53 Furthermore, L-proline-catalysed aldol reactions, which are related to the Knoevenagel condensation, have been shown to involve the activation of aldehyde as an enamine.54–61 Lewis acids such as Zn(ClO)4·6H2O also catalyse the formation of β-enaminoesters from β-ketoesters and primary or secondary amines.62,63 Therefore, the activation of methyl acetoacetate in the form of β-enaminoester 11 in addition to the enolate ion 10 can be proposed. Although 11 was not detected in the reaction mixture, its formation as a transient species cannot be ruled out. The generation of Knoevenagel adduct-Eu(III) complex (15) is consistent with the well-known ability of lanthanide ions to chelate β-dicarbonyl compounds.64 Azafulvenium salt 18 could not be isolated due to its high instability.65,66 However, its formation is proved by the isolation of bis-indole derivative 19, which can only come from 18 by addition of a second molecule of indole. The formation of 3,3′-bis-indolylmethanes from indole and carbonyl compounds is catalysed by Lewis acids67–71 as well as strong Brønsted acids.65,66 Because L-proline is a weak Brønsted acid, its catalytic effect in the mechanism of Scheme 2 is expected to be negligible with respect to that of europium ion. The role of solvent can be rationalised in the light of proposed mechanisms as well. Alcohols can stabilize several reactive intermediates through specific interactions: intermediates 10 and 12 (by H-bond donation), 15, 16, and 17 (by complexation of Eu3+ ion), and 18 (by H-bond acceptance from the indolic NH group). Alcohols, being polar protic solvents, can also catalyse proton transfer reactions involved in the formation of intermediates 10–13, 17, 18 and 20, thus favouring the overall reaction. In the light of the evidence reported above, both mechanisms seem to take place simultaneously, although data do not allow to establish which one is prevalent.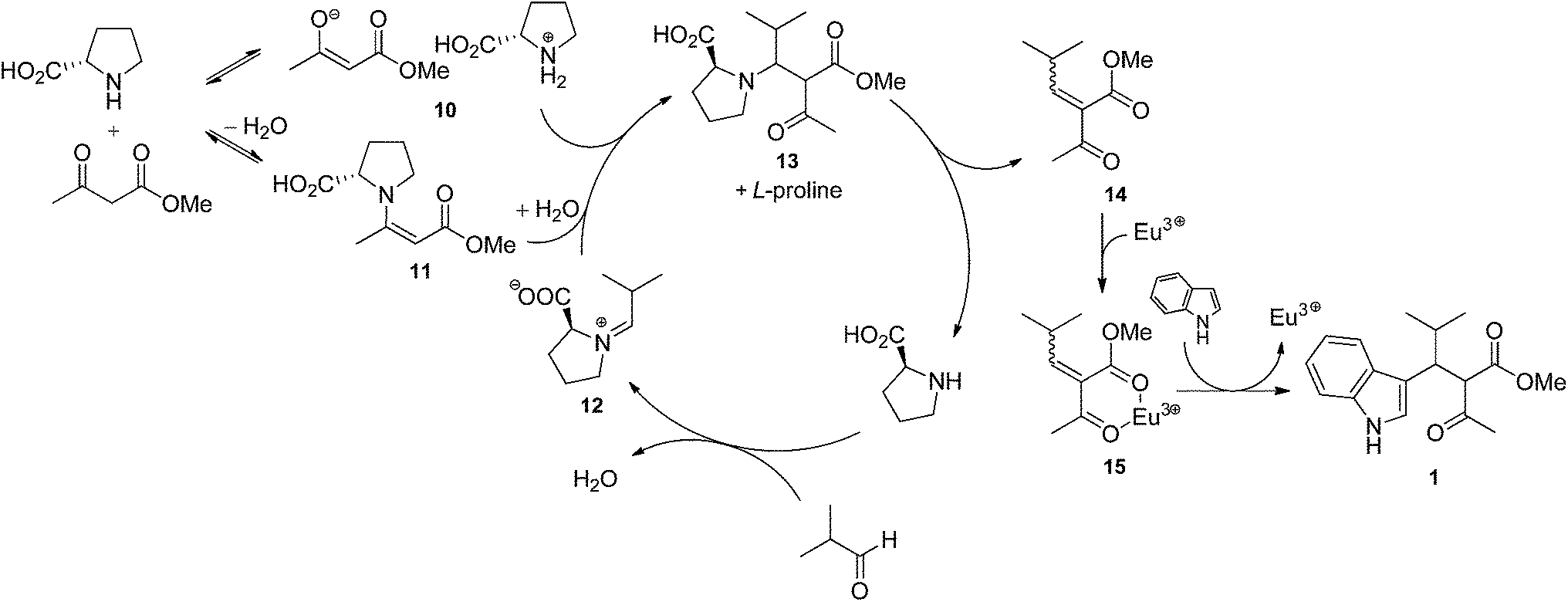 | ||
| Scheme 1 Mechanism via Knoevenagel adduct. | ||
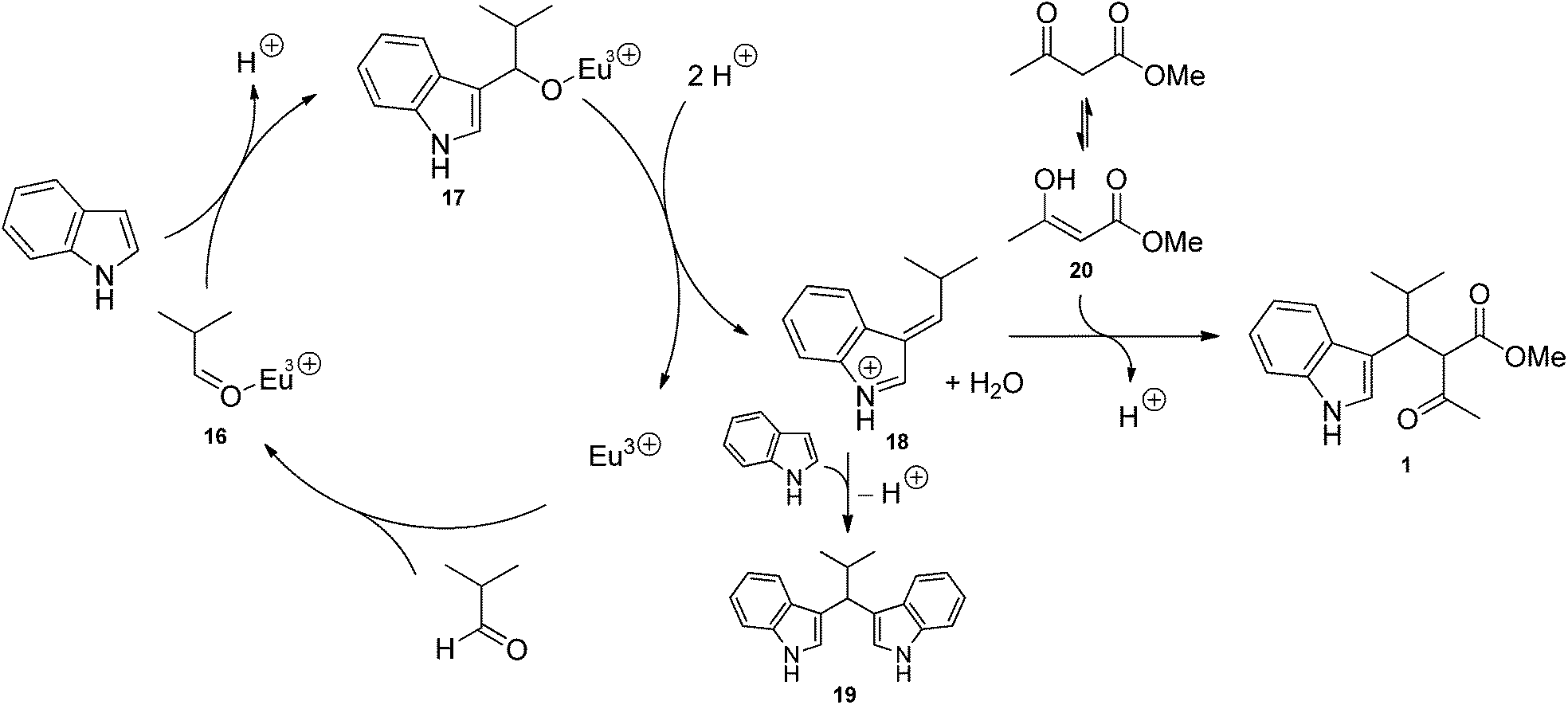 | ||
| Scheme 2 Mechanism via azafulvenium ion. | ||
2.5 Diastereoselectivity
Unlike chemical yield, diastereoselectivity was high, as most compounds with two stereocenters (1, 2, 3, 5, and 8) were isolated as a single diastereomer. Stereochemistry of 1 and 3 has previously been determined.6 In principle, the observed diastereoselectivity can be explained in two ways. The first possibility is that diastereoselectivity is induced by L-proline through the formation of a Knoevenagel adduct-Eu(III)-proline complex. In such a complex, L-proline would fold back and cover one face of the adduct, thus forcing indole to attack the opposite face. Protonation of the resulting intermediate on the same side with respect to proline would lead to the condensation product diastereoselectively. The formation of Eu(III)-proline complex is reasonable in the light of the high oxophilicity of lanthanide ions as well as the chelating ability of α-amino acids; Eu(III)-proline complexes are actually known, both in solid phase72 and in solution.73,74 Nevertheless, experimental evidences allowed us to rule out this hypothesis. As a matter of fact, products 1 and 3 were obtained as a single diastereomer in the presence of both optically pure and racemic proline or even without proline in methanol (Table 3, entry 1). These results show that L-proline is not involved in the stereochemical outcome of the reaction.75 Another more likely explanation for the observed diastereoselectivity is that it is induced by crystallisation.76,77 The condensation product bears an enolisable hydrogen on the α-carbon, so the two diastereomers may interconvert via enolisation. If one diastereomer crystallises, it is subtracted from the reaction mixture, and the interconversion equilibrium is continuously displaced towards the crystalline form until the whole compound is present as one pure diastereomer (crystallisation-induced asymmetric transformation). Based on this model, diastereoselectivity is depending on the intrinsic solubility properties of diastereomers. The hypothesis of diasteromeric interconversion is supported by the fact that some active methylene compounds such as α-nitroketones78 have an enantiomerisation barrier low enough to allow a rapid inversion of their stereogenic centre at room temperature. In addition, crystallisation-induced asymmetric transformation of diastereomers has been reported in several reactions,79,80 including multicomponent reactions.81,82 Obviously, this type of transformation depends solely on the relative thermodynamic stability of the corresponding crystal lattices and may only take place if at least one diastereomer is solid. If both diastereomers crystallise from the reaction medium (as in the case of 6a and 6b) no diastereoselectivity is observed.2.6 Greenness
The reaction developed in this work adheres to several principles of green chemistry: (1) it is one-pot; (2) it is catalytic; (3) it uses safe catalysts (a natural amino acid and a water-stable salt); (4) it runs at room temperature; and (5) it takes place in a green solvent (methanol). Reaction time, although long (6 days), is comparable to that of some green reactions such as the polyphosphate-induced formation of peptides in water (10–15 days).83 Because the reaction we developed is supposed to be green, we wondered how green it is. One of the most common metrics to measure reaction “greenness” or cleanliness is the environmental factor (E-factor),84,85 defined as the ratio of waste mass over product mass. E-factor provides a realistic evaluation of the actual amount of waste produced as it takes into account the reaction yield as well as all reactants and solvents involved in the reaction, including those necessary for work-up. E-factor calculated for the reaction of indole, isobutyraldehyde, and methyl acetoacetate catalysed by Eu(OTf)3 and proline in methanol is 0.70 (Table S4†). In other words, the reaction produces 0.70 kg of waste per kg of product. Considering that typical E-factors range from 0.01 (for the cleanest reactions) to more than 100 (for the dirtiest ones),86 the Eu(III)/proline-catalysed reaction is quite clean. As a matter of comparison, E-factor for the Ti(IV)-promoted reaction is 3.40. This means that the catalytic reaction produces about five times less waste than the stoichiometric reaction.3 Conclusions
In conclusion, we have developed a Yonemitsu-type trimolecular condensation of aromatic heterocycles, aldehydes, and active methylene compounds catalysed by L-proline and europium triflate. The importance of this study is twofold. From a synthetic viewpoint, the reaction can find application in the synthesis of polyfunctionalised heterocycles. From a methodological viewpoint, the reaction respects a number of green chemistry's principles, and works with substrates that are unreactive under classical conditions, in some cases with high diastereoselectivity.This study showed how to convert a non-green reaction into a green reaction in a rational way based on its reaction mechanism, and hopefully will pave the way to the development of green protocols for other reactions.
Acknowledgements
This work was carried out with support from the University ‘G. d’Annunzio’ of Chieti-Pescara, the University of Rome, and MIUR (PRIN 2010–11, prot. 2010N3T9M4). A. R. thanks the Japan Society for the Promotion of Science for providing a JSPS Invitation Fellowship for Research in Japan (FY2013).Notes and references
- M. Lancaster, Green Chemistry: An Introductory Text, Royal Society of Chemistry, 2002 Search PubMed.
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford Univ Press, 1998 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- K. Sanderson, Nature, 2011, 469, 18–20 CrossRef CAS PubMed.
- A. Renzetti, E. Dardennes, A. Fontana, P. De Maria, J. Sapi and S. Gérard, J. Org. Chem., 2008, 73, 6824–6827 CrossRef CAS PubMed.
- S. Gérard, A. Renzetti, B. Lefevre, A. Fontana, P. De Maria and J. Sapi, Tetrahedron, 2010, 66, 3065–3069 CrossRef PubMed.
- L. A. Marcaurelle and C. W. Johannes, Prog. Drug Res., 2008, 66, 187–216 Search PubMed.
- A. Marrone, A. Renzetti, P. De Maria, S. Gérard, J. Sapi, A. Fontana and N. Re, Chem.–Eur. J., 2009, 15, 11537–11550 CrossRef CAS PubMed.
- S. Tascioglu, Tetrahedron, 1996, 52, 11113–11152 CrossRef CAS.
- A. Chatterjee, S. K. Hota, M. Banerjee and P. K. Bhattacharya, Tetrahedron Lett., 2010, 51, 6700–6703 CrossRef CAS PubMed.
- E. E. Coyle, K. Joyce, K. Nolan and M. Oelgemoeller, Green Chem., 2010, 12, 1544–1547 RSC.
- R. Jain, K. Sharma and D. Kumar, Tetrahedron Lett., 2012, 53, 6236–6240 CrossRef CAS PubMed.
- H. Matondo, J. C. Garrigues, I. Rico-Lattes and A. Lattes, Appl. Organomet. Chem., 2009, 23, 191–195 CrossRef CAS.
- H. Yao and D. E. Richardson, J. Am. Chem. Soc., 2003, 125, 6211–6221 CrossRef CAS PubMed.
- P. De Maria, A. Fontana, C. Gasbarri and G. Siani, Tetrahedron, 2005, 61, 7176–7183 CrossRef CAS PubMed.
- P. De Maria, A. Fontana and G. Cerichelli, J. Chem. Soc., Perkin Trans. 2, 1997, 2329–2334 RSC.
- S. Guernelli, R. Noto, C. Sbriziolo, D. Spinelli and M. L. T. Liveri, J. Colloid Interface Sci., 2001, 239, 217–221 CrossRef CAS PubMed.
- S. Guernelli, A. Fontana, R. Noto, D. Spinelli and L. M. L. Turco, J. Colloid Interface Sci., 2012, 381, 67–72 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- C. Capello, U. Fischer and K. Hungerbuehler, Green Chem., 2007, 9, 927–934 RSC.
- C. Jimenez-Gonzalez, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 2005, 7, 42–50 CrossRef CAS.
- J. Ren, C. J. Cramer and R. R. Squires, J. Am. Chem. Soc., 1999, 121, 2633–2634 CrossRef CAS.
- T. Ollevier and E. Nadeau, Org. Biomol. Chem., 2007, 5, 3126–3134 CAS.
- T. Ollevier, Top. Curr. Chem., 2012, 311, 69–114 CrossRef CAS.
- S. Repichet, A. Zwick, L. Vendier, R. C. Le and J. Dubac, Tetrahedron Lett., 2002, 43, 993–995 CrossRef CAS.
- S. Kobayashi and C. Ogawa, Chem.–Eur. J., 2006, 12, 5954–5960 CrossRef CAS PubMed.
- Y. Oikawa, H. Hirasawa and O. Yonemitsu, Tetrahedron Lett., 1978, 1759–1762 CrossRef CAS.
- Toluene, although not an eco-friendly compound, was tested to evaluate the feasibility of reaction in non-polar solvents.
- P. Amrhein and K. Rück-Braun, in Organic Synthesis Highlights IV, Wiley-VCH Verlag GmbH, 2008, pp. 104–109 Search PubMed.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W. L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed.
- W. Xie, Y. Jin and P. G. Wang, CHEMTECH, 1999, 29, 23–29 CAS.
- S. Kobayashi, in Organic Synthesis in Water, Springer, Netherlands, 1998, pp. 262–305 Search PubMed.
- B. O. Okandeji, J. R. Gordon and J. K. Sello, J. Org. Chem., 2008, 73, 5595–5597 CrossRef CAS PubMed.
- S. Kobayashi, S. Komiyama and H. Ishitani, Biotechnol. Bioeng., 1998, 61, 23–31 CrossRef CAS.
- Preliminary results of this work have previously been presented. Please see: (a) A. Renzetti, A. Fontana, S. Gérard, J. Sapi and P. De Maria, How to make the trimolecular condensation of indole, isobutyraldehyde and methyl acetoacetate green and catalytic (poster), 42nd IUPAC World Chemistry Congress, Glasgow (UK), August 2–7 2009 Search PubMed; (b) A. Renzetti, M. Colazzo, E. Boffa, S. Gérard, J. Sapi, T.-H. Chan and A. Fontana, Green Yonemitsu-type trimolecular condensations (poster), 18th European Symposium on Organic Chemistry, Marseille (France), July 7–12 2013 Search PubMed.
- F. Epifano, S. Genovese, O. Rosati, S. Tagliapietra, C. Pelucchini and M. Curini, Tetrahedron Lett., 2011, 52, 568–571 CrossRef CAS PubMed.
- J. Zhou and Y. Tang, J. Am. Chem. Soc., 2002, 124, 9030–9031 CrossRef CAS PubMed.
- C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 107–163 Search PubMed.
- G. Angelini, C. Chiappe, P. De Maria, A. Fontana, F. Gasparrini, D. Pieraccini, M. Pierini and G. Siani, J. Org. Chem., 2005, 70, 8193–8196 CrossRef CAS PubMed.
- Z. Rappoport, The Chemistry of Enols, John Wiley & Sons, 1990 Search PubMed.
- A. Renzetti, PhD Thesis, Università di Chieti-Pescara and Université de Reims Champagne-Ardenne, 2007.
- P. E. Harrington and M. A. Kerr, Synlett, 1996, 1047–1048 CrossRef CAS PubMed.
- J. S. Yadav, S. Abraham, B. V. S. Reddy and G. Sabitha, Synthesis, 2001, 2001, 2165–2169 CrossRef.
- S.-J. Ji and S.-Y. Wang, Synlett, 2003, 2003, 2074–2076 CrossRef.
- Z.-P. Zhan, R.-F. Yang and K. Lang, Tetrahedron Lett., 2005, 46, 3859–3862 CrossRef CAS PubMed.
- Z.-H. Huang, J.-P. Zou and W.-Q. Jiang, Tetrahedron Lett., 2006, 47, 7965–7968 CrossRef CAS PubMed.
- R. Pal, A. Das Gupta and A. K. Mallik, ISRN Org. Chem., 2012, 2012, 674629 Search PubMed.
- W. Zhuang, T. Hansen and K. A. Jorgensen, Chem. Commun., 2001, 347–348 RSC.
- Y.-Y. Zhou, X.-L. Sun, B.-H. Zhu, J.-C. Zheng, J.-L. Zhou and Y. Tang, Synlett, 2011, 2011, 935–938 CrossRef PubMed.
- D. A. Evans, K. R. Fandrick, H.-J. Song, K. A. Scheidt and R. Xu, J. Am. Chem. Soc., 2007, 129, 10029–10041 CrossRef CAS PubMed.
- A. Erkkilae, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef CAS PubMed.
- L. F. Tietze and U. Beifuss, in Comprehensive Organic Synthesis, ed. M. T. Barry and F. Ian, Pergamon, Oxford, 1991, pp. 341–394 Search PubMed.
- S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 11273–11283 CrossRef CAS PubMed.
- M. Arno and L. R. Domingo, Theor. Chem. Acc., 2002, 108, 232–239 CrossRef CAS PubMed.
- L. Hoang, S. Bahmanyar, K. N. Houk and B. List, J. Am. Chem. Soc., 2003, 125, 16–17 CrossRef CAS PubMed.
- F. R. Clemente and K. N. Houk, Angew. Chem., Int. Ed., 2004, 43, 5766–5768 CrossRef CAS PubMed.
- J. Kofoed, T. Darbre and J.-L. Reymond, Chem. Commun., 2006, 1482–1484 RSC.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Angew. Chem., Int. Ed., 2010, 49, 4997–5003 CrossRef CAS PubMed.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, J. Org. Chem., 2011, 76, 3005–3015 CrossRef CAS PubMed.
- M. B. Schmid, K. Zeitler and R. M. Gschwind, Chem.–Eur. J., 2012, 18, 3362–3370 CrossRef CAS PubMed.
- G. Bartoli, M. Bosco, M. Locatelli, E. Marcantoni, P. Melchiorre and L. Sambri, Synlett, 2004, 239–242 CrossRef CAS PubMed.
- A. S. Demir and M. Emrullahoglu, Tetrahedron, 2006, 62, 1452–1458 CrossRef CAS PubMed.
- N. K. Dutt, S. Sur and S. Rahut, J. Inorg. Nucl. Chem., 1971, 33, 1717–1724 CrossRef CAS.
- R. A. Jones, in Comprehensive Heterocyclic Chemistry, ed. R. K. Alan and W. R. Charles, Pergamon, Oxford, 1984, pp. 201–312 Search PubMed.
- G. F. Smith, Adv. Heterocycl. Chem., 1963, 18, 287–309 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor and A. A. Jafari, J. Mol. Catal. A: Chem., 2006, 244, 168–172 CrossRef CAS PubMed.
- M. Hosseini-Sarvari, Synth. Commun., 2008, 38, 832–840 CrossRef CAS.
- S.-J. Ji, M.-F. Zhou, D.-G. Gu, S.-Y. Wang and T.-P. Loh, Synlett, 2003, 2077–2079 CrossRef CAS PubMed.
- R. Nagarajan and P. T. Perumal, Tetrahedron, 2002, 58, 1229–1232 CrossRef CAS.
- Z.-H. Zhang, L. Yin and Y.-M. Wang, Synthesis, 2005, 1949–1954 CAS.
- E. Huskowska, I. Turowska-Tyrk, J. Legendziewicz and T. Glowiak, J. Alloys Compd., 1998, 275–277, 852–858 CrossRef CAS.
- X. Hu, Z. Shan and C. Qing, Lett. Org. Chem., 2012, 9, 493–496 CrossRef CAS.
- S. Lal, Aust. J. Chem., 1972, 25, 1571–1573 CrossRef CAS.
- The only advantage in using optically active proline is that L-proline is a natural amino acid, and as such is much cheaper than the D enantiomer or the racemate.
- E. L. Eliel, Elements of Stereochemistry, John Wiley & Sons, 1969 Search PubMed.
- J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates, and Resolutions, John Wiley & Sons, 1981 Search PubMed.
- F. Gasparrini, M. Pierini, C. Villani, P. De Maria, A. Fontana and R. Ballini, J. Org. Chem., 2003, 68, 3173–3177 CrossRef CAS PubMed.
- N. G. Anderson, Org. Process Res. Dev., 2005, 9, 800–813 CrossRef CAS.
- R. Yoshioka, Top. Curr. Chem., 2007, 269, 83–132 CrossRef CAS.
- W. H. J. Boesten, J.-P. G. Seerden, L. B. de, H. J. A. Dielemans, H. L. M. Elsenberg, B. Kaptein, H. M. Moody, R. M. Kellogg and Q. B. Broxterman, Org. Lett., 2001, 3, 1121–1124 CrossRef CAS PubMed.
- P. Jakubec, P. Petráš, A. Ďuriš and D. Berkeš, Tetrahedron: Asymmetry, 2010, 21, 69–74 CrossRef CAS PubMed.
- J. Rabinowitz and A. Hampai, J. Mol. Evol., 1984, 21, 199–201 CrossRef.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization data for all compounds, and Tables S1–S4. See DOI: 10.1039/c4ra08853k |
| This journal is © The Royal Society of Chemistry 2014 |