
Catalytic dehydrogenation of isobutane over Co-based catalysts†
Guowei Wanga,
Xiaolin Zhuab,
Jiaoyu Zhanga,
Yanan Suna,
Chunyi Li*a and
Honghong Shana
aState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266580, China. E-mail: chyli_upc@126.com; chyli@upc.edu.cn; Fax: +86 532 86981718; Tel: +86 532 86981862
bBeijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China
First published on 28th October 2014
Abstract
In this work, to optimize the catalytic performance of Co-based catalysts for isobutane dehydrogenation, the effects of support, calcination temperature and some promoters were investigated systematically. Results of activity tests and catalyst characterization jointly indicated that both the support and calcination temperature influenced Co dispersion, catalyst acid properties and the interaction between support and Co species significantly. Consequently, the adsorption–desorption behaviors for isobutane and isobutene as well as the dehydrogenation performance were further affected. Sulfided Co/SiO2, with no acidity, exhibited the best performance. Although high calcination temperature was beneficial for achieving a high selectivity to isobutene over Al2O3 and MgAl2O4 supported Co-based catalysts, the inevitable formation of Co2SiO4 over Co/SiO2 at high temperature led to reduced active sites and dehydrogenation activity. In addition, by comparing the different effects of the separate introduction of S, Sn, Cu and Cl, it was concluded that an efficient promoter should present the following characteristics: inhibition of the formation of metal ensembles, and strong binding affinity to two hydrogen atoms of one additive atom.
1. Introduction
Lately, dehydrogenation of light alkanes has received great attention as a route to produce the corresponding alkenes, which constitute an important and versatile class of chemical intermediate. However, due to the high cost of platinum and adverse environment effects of chromia,1 the application of industrialized Pt and Cr2O3-based catalysts has been limited to some extent.Although high catalytic activity can be obtained over metal (such as Fe, Co and Ni) based catalysts for the conversion of isobutane, the reaction is suffered from an extremely poor selectivity because of aggravated hydrogenolysis reaction, of which the main product is methane rather than isobutene.2,3 To address this issue, sulfur was introduced into metal based catalysts in spite that it is usually regarded as an impurity.4–6 Fortunately, we have found that a series of metal sulfide catalysts, either prepared by impregnation with sulfate7–9 or sulfidation with H2S,3,10 exhibit excellent performance in alkane dehydrogenation. The conversion of isobutane over a series of sulfide catalysts of Fe, Co, and Ni reached up to 70 wt%, with a selectivity to isobutene of around 87 wt%. In general, the promoting effects of introduced sulfur can be understood in two aspects:3 one is a geometric effect to dilute aggregated metallic species and inhibit C–C bond rupture; another is an electronic effect to facilitate olefin desorption and further increase product selectivity. Meanwhile, sulfur loss is speculated to be the origin of catalyst deactivation. To replenish the loss of sulfur, continuous sulfidation-reaction cycles have been carried out and the catalyst performance can be maintained at a high level. Moreover, it should also be noted that, the catalytic performance can be stable for a long period when diluted H2S is fed together with isobutane.10
As mentioned above, the active sites of metal sulfide catalysts and how they interact with isobutane molecules were speculated in our previous study. However, it is still not enough for the preparation of an ideal dehydrogenation catalyst. Firstly, the catalyst support, significantly influencing the dispersion and electronic properties of the active components11,12 as well as the acid–base properties of the catalysts,13–15 is an important factor to be considered. Meanwhile, some literature reported that high-temperature calcination could modify the catalyst structure and increase the activity and stability of Pd@CeO2/Si–Al2O3 catalyst obviously.16 Therefore, the effects of both support and calcination condition, directly related to catalyst structure16–18 and properties19,20 as well as the interaction between active component and support,21,22 should be further investigated.
Given the difference in acid–base properties and the interaction with active component, Al2O3, MgAl2O4 and SiO2 were chosen as the supports of Co-based catalysts separately in this work. Moreover, to examine the effect of calcination temperature, the catalysts with different supports were calcined at both low and high temperatures.
In addition to the support and calcination temperature, the promoter plays a more important role in this catalyst system. As reported in our previous study, metal ensembles catalyze hydrogenolysis reaction to generate methane. Since the hydrogenolysis reaction is structure sensitive,23–27 breaking these metal ensembles and isolating small metal particles seem to be the key in restricting this reaction. To achieve this goal, there are two available methods: reducing metal loading or introducing barriers into the ensembles. As for the former case, although the selectivity to isobutene increased obviously with decreasing metal loading, the catalyst activity decreased sharply,7 indicating the lack of active sites. In contrast, the later approach seems to be more effective: sulfur introduction inhibits the formation of metal ensembles and improves the dehydrogenation performance significantly. Similarly, Apesteguía and Barbier28 also pointed out that presulfidation treatment of Pt-based catalyst could inhibit hydrogenolysis reaction effectively.
Some open literatures reported that the addition of Sn into Pt-based catalyst generated certain geometric and electronic effects, which effectively reduced platinum particles and suppressed coke formation and hydrogenolysis reaction.29–32 Similarly, copper addition into nickel-based catalyst also played such a role through the formation of Cu–Ni alloys.33,34 In addition, resulting from blockage/electronic effects of the adatom, chlorine modification influenced the adsorptive and catalytic properties of the catalyst remarkably.35–37 Therefore, to further explore the promoting mechanism of the additives in this work, Sn, Cu, and Cl have been introduced into Co/SiO2 catalyst to compare their effects with that of S on the dehydrogenation performance.
2. Experimental
2.1 Catalyst preparation
Alumina was prepared by the sol–gel method. Firstly, pseudo-boehmite powder was mixed with distilled water to obtain a suspended solution. After that, HCl solution (36–38 wt%) was added dropwise under vigorous stirring at 70 °C, until a gel-like mixture was formed. To prepare magnesia-alumina spinel, an appropriate amount of magnesium nitrate solution was added into the above gel-like mixture. Then, the mixture was stirred for 2 h, dried at 140 °C for 12 h, and calcined at 700 °C for another 2 h in air. Finally, both gel-like mixtures were crushed and sieved to 80–180 μm for later use. In addition, mesoporous silica (80–180 μm) was obtained from China Qingdao Haiyang Chemical Company, with a specific surface area of 378.3 m2 g−1 and an average pore diameter of 9.6 nm.Supported catalyst samples were prepared by wetness impregnation of the support with certain amount of cobalt nitrate solution (Co3O4 content of the catalyst was 13 wt% in this work). Subsequently, the samples were dried overnight at 140 °C and calcined for 2 h at different temperatures. The as-prepared samples are denoted as 13Co/support-x, where “x” indicated the calcination temperature. Moreover, sulfided catalysts were obtained by pretreatment of the samples with H2S/H2 (30 mL min−1) for 3 h at 560 °C. Co/SiO2 catalysts with the introduction of Sn or Cu were prepared by subsequent impregnation of above as-prepared Co/SiO2 catalysts with ethanol solution of stannous chloride or aqueous solution of copper nitrate, respectively. Cl-modified catalyst was prepared by impregnation of SiO2 with certain amount of cobalt chloride solution. After the introduction of the promoters, the catalysts were calcined again at 560 °C for 2 h.
2.2 Catalyst characterization
Nitrogen adsorption–desorption measurements were performed on a Quadrasorb SI instrument and a porosimetry analyzer at liquid nitrogen temperature. The specific surface areas and pore volumes of the catalyst samples were determined by the BET and BJH methods. X-ray diffraction (XRD) analysis of the samples was carried out on an X'Pert PRO MPD diffractometer system using Cu Kα radiation at 40 kV and 40 mA, running from 5° to 75° with scanning speed at 10° min−1. To determine the content of chlorine on 13Co/SiO2–Cl catalyst before and after reaction, elemental analysis was carried out by an Axios X-ray flourescence spectrometer (XRF).Metal dispersion and particle size were determined by H2 pulse chemisorption using a FINESORB-3010 chemisorption analyzer, with the gas released from the reactor being analyzed by an online GAS-100Q quadrupole mass spectrometer. About 0.3 g of fresh catalyst was reduced in situ under a flow of 10 vol% H2/N2 (30 mL min−1) at 773 K for 1 h, and then cooled down to 100 °C under the purge of Ar. Afterwards, H2/N2 was pulsed into the catalyst bed by the application of a six-way valve until the signal intensity of H2 did not increase with the pulse number any more. Ultimately, according to the change of area for each pulse, the amount of chemisorbed H2 was determined, and thus the metal dispersion and particle size can be further calculated.
To investigate the desorption behaviors of the catalysts for alkenes, isobutene desorption test was performed on the same chemisorption analyzer, also equipped with an online MS. For each test, about 0.3 g of sulfided catalyst was loaded into a quartz tube reactor, preheated in a flow of nitrogen (60 mL min−1) at 450 °C for 0.5 h, and then cooled down to 200 °C. Subsequently, with the application of a four-way valve, nitrogen flow was instantaneously switched to a mixture of Ar and isobutene at separate flow rate of 30 mL min−1. After adsorption saturation, the valve was switched back to nitrogen. At that time, the signal for the inert gas decreased immediately since it is hardy adsorbed on catalyst surface. However, the decrease of signal for isobutene was delayed due to its adsorption on the catalyst. Precisely the difference between the signals of inert gas and isobutene gives useful desorption information, for instance, a larger delay extent is indicative of a more difficult isobutene desorption process. Furthermore, such test was also performed at 300 °C and 400 °C.
To compare the adsorption amounts of isobutane or isobutene over different catalysts, pulse chemisorption tests were also carried out using the same equipment with H2 chemisorption. After pretreatment of 0.3 g of sulfide catalysts in a flow of nitrogen, 0.803 mL of isobutane or isobutene was pulsed into the catalyst bed with a flow of nitrogen (30 mL min−1) at 300 °C. Meanwhile, an online quadrupole mass spectrometer was applied to detect the signals of released hydrocarbons, and the adsorption amount could be further determined by calculating the signal area of each pulse.
The acidic properties of the catalyst samples were determined by temperature-programmed desorption (TPD) of NH3. For each test, 0.1 g of sample was preheated in situ under a helium flow at 600 °C for 0.5 h, and then cooled down to 100 °C. Adsorption was proceeded for 0.5 h under NH3 flow. Subsequently, the sample was purged under a dry helium flow to remove physically adsorbed NH3. Ultimately, desorption was initiated at a heating rate of 10 °C min−1 until 600 °C was reached. The amount of desorbed NH3 was calculated from the area under the TPD curve.
2.3 Catalytic activity test
Catalytic activity tests were performed in a fixed bed micro-reactor at atmospheric pressure and 560 °C. Prior to reaction, S-modified catalysts were obtained by in situ sulfidation with H2S/H2 (30 mL min−1) for 3 h at 560 °C, while Sn, Cu, and Cl-promoted catalysts were reduced by H2 (30 mL min−1) for 2 h at 560 °C. During the test, 4 g of catalyst was loaded into the reactor, then a flow of isobutane diluted by nitrogen with a fixed molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 was fed into the reactor, at a total flow rate of 14 mL min−1. The inner diameter of the reactor is 14 mm, with a length of 160 mm, and the catalyst bed (50 mm long) was loaded between two layers of silica sand. To compare the dehydrogenation performance of different catalysts, the measurements were taken at the initial stage of the reaction. Both the feed and products were analyzed by a Bruker 450 Gas Chromatograph (GC) equipped with an FID detector and two TCD detectors.
:6 was fed into the reactor, at a total flow rate of 14 mL min−1. The inner diameter of the reactor is 14 mm, with a length of 160 mm, and the catalyst bed (50 mm long) was loaded between two layers of silica sand. To compare the dehydrogenation performance of different catalysts, the measurements were taken at the initial stage of the reaction. Both the feed and products were analyzed by a Bruker 450 Gas Chromatograph (GC) equipped with an FID detector and two TCD detectors.
Furthermore, isobutane dehydrogenation in a pulse mode fixed-bed reactor over Cu-modified catalysts was also conducted. For each test, about 0.1 g of catalyst was loaded and the reaction temperature was maintained at 560 °C. Prior to the reaction, the catalyst was pretreated by a flow of H2/N2 (30 mL min−1) for 2 h. Then, pulse feeding was performed using a six-way valve, and 0.803 mL of isobutane was driven into the reactor by nitrogen flow (with a flow rate of 30 mL min−1) in one pulse. Meanwhile, the effluent products were characterized using an online MS, and their mass peaks were registered as follows: H2, 2; CH4, 16; i-C4H8, 56 and i-C4H10, 58.
3. Results and discussion
3.1 Structural characterization and Co dispersion
The XRD patterns of Co-based catalysts supported on the three different supports are illustrated in Fig. 1a. For low-temperature calcined catalysts, diffraction peaks characteristic of both the support and Co3O4 were observed. However, with the increase of calcination temperature, some newly emerged diffraction peaks assigned to CoAl2O4 and Co2SiO4 were detected over Co/Al2O3 and Co/SiO2 catalysts, respectively. Meanwhile, the intensities of peaks corresponding to Co3O4 decreased to some extent. However, no obvious phase change took place over Co/MgAl2O4 catalyst with increasing calcination temperature.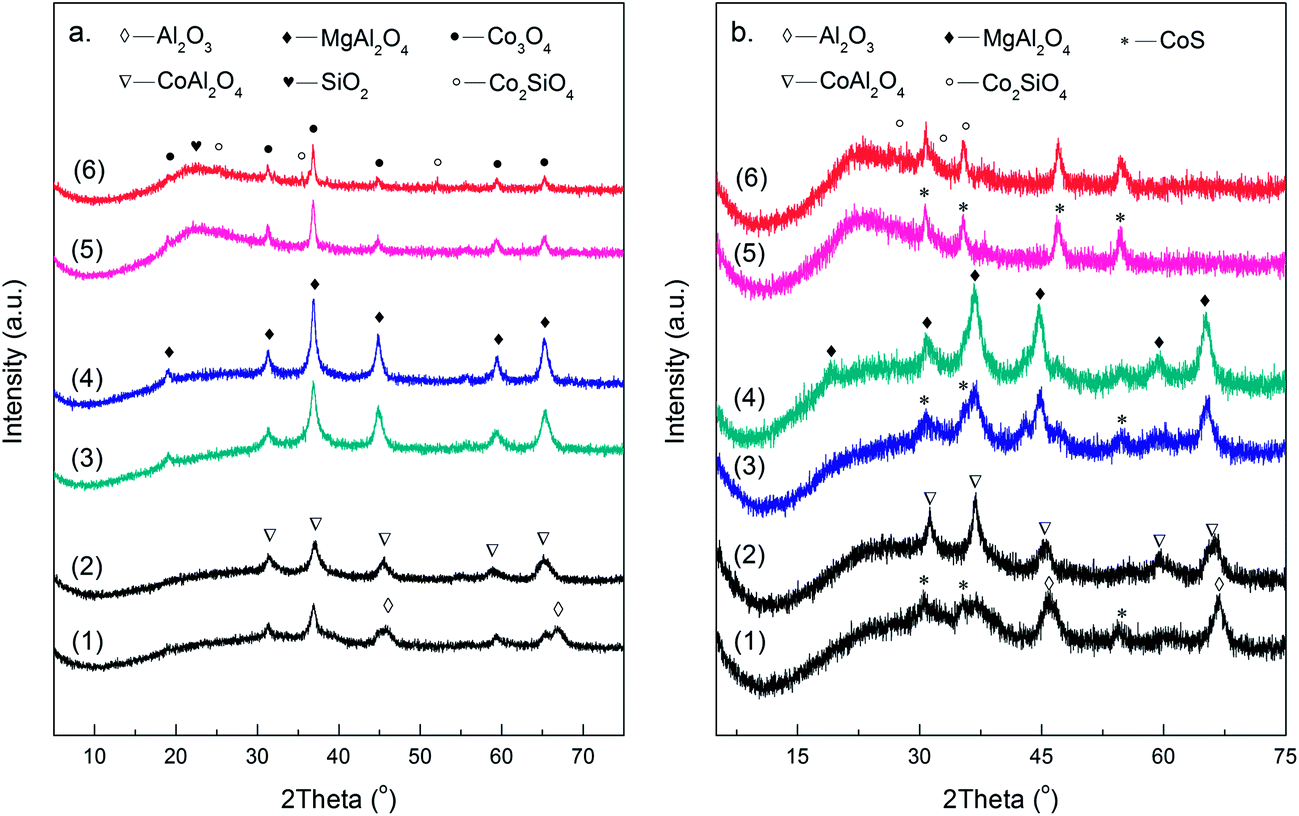 | ||
| Fig. 1 XRD patterns of different Co-based catalysts before (a) and after (b) sulfidation: (1) 13Co/Al2O3-560; (2) 13Co/Al2O3-800; (3) 13Co/MgAl2O4-560; (4) 13Co/MgAl2O4-800; (5) 13Co/SiO2-560; (6) 13Co/SiO2-800. | ||
Moreover, phase analysis was further carried out over sulfided Co-based catalysts with different supports calcined at both low and high temperatures, and the XRD patterns are shown in Fig. 1b. CoS was present on the surface of catalysts calcined at 560 °C after sulfidation, indicating that sulfidation treatment transformed Co3O4 into CoS successfully. However, when the calcination temperature increased to 800 °C, stable CoAl2O4 and Co2SiO4, which were difficult to be sulfided, formed on the surface of Co/Al2O3 and Co/SiO2 catalysts, respectively. In addition, compared with low temperature calcined samples, the intensity of diffraction peaks corresponding to CoS decreased to some extent. As for MgAl2O4-supported catalyst, no obvious phase change was observed at high calcination temperature.
Table 1 lists the BET surface area, pore properties and Co dispersion determined by H2 chemisorption of Co-based catalysts calcined at different temperatures. With the increase of calcination temperature, the surface area decreased noticeably for all the samples, which was indicative of the collapse of small pore walls. And this result was also consistent with the increasing trend of pore size. As for the catalysts with different supports, the surface area decreased in the following order: 13Co/SiO2 > 13Co/Al2O3 > 13Co/MgAl2O4. While, for the sequence of surface density of Co atom, a reverse trend to that of surface area was obtained. Furthermore, Co dispersion was determined by H2 chemisorption, and it decreased with calcination temperature. Meanwhile, Co dispersion increased as follows: 13Co/SiO2 < 13Co/MgAl2O4 < 13Co/Al2O3, and the calculated particle size of Co presented an opposite trend. Above results indicated that the dispersion of Co species had no direct relationship with the catalyst surface area, and the interaction between Co species and the support might play a more significant role.
| Sample | SBETa, m2 g−1 | Vpb, cm3 g−1 | Dpc, nm | Cosurf. density, atomCo/nm2 | Co dispersiond, % | DCoe, nm | TOFf, h−1 |
|---|---|---|---|---|---|---|---|
| a Specific surface area.b Pore volume.c Pore size.d Co dispersion was determined by H2 chemisorption, referred to the molar ratio of surface Co to bulk Co, with the latter being calculated from the cobalt nitrate impregnation.e Particle size of Co was determined by H2 chemisorption.f Turn over frequency was calculated as the amount of formed isobutene per hour divided by the amount of active metal atom (determined by H2 chemisorption) supported on catalyst after presulfidation treatment with H2S/H2 (30 mL min−1) for 3 h at 560 °C. | |||||||
| 13Co/Al2O3-560 | 146 | 0.25 | 5.7 | 6.7 | 43.6 | 2.3 | 0.4 |
| 13Co/Al2O3-800 | 130 | 0.25 | 6.5 | 7.5 | 31.2 | 3.1 | 0.7 |
| 13Co/MgAl2O4-560 | 84 | 0.19 | 7.9 | 11.6 | 28.5 | 3.4 | 1.2 |
| 13Co/MgAl2O4-800 | 73 | 0.19 | 7.9 | 13.4 | 23.4 | 4.2 | 1.9 |
| 13Co/SiO2-560 | 225 | 0.86 | 8.7 | 4.3 | 20.3 | 4.3 | 2.5 |
| 13Co/SiO2-800 | 168 | 0.73 | 9.6 | 5.8 | 11.5 | 8.5 | 3.9 |
3.2 Effects of support and calcination temperature
The influence of calcination temperature on dehydrogenation performance of sulfided Co-based catalysts was firstly investigated, and the results are illustrated in Fig. 2. With regard to Al2O3-supported catalyst, isobutane conversion decreased obviously from 76.2 wt% at 560 °C to 57.1 wt% at 800 °C. Meanwhile, the selectivity to methane and n-butane also exhibited a gradually decreasing trend, indicating that both cracking and isomerization reactions were weakened with increasing calcination temperature. Even though, the selectivity to isobutene only increased to 46.6 wt%, a relatively low level. As for 13Co/MgAl2O4 catalyst, the selectivity to isobutene was much higher, and it increased from 60.4 wt% to 84.0 wt% with the increase of calcination temperature. Meanwhile, isobutane conversion declined slightly at the initial stage, and then approached constant afterwards. When taking SiO2 as the support, the selectivity to isobutene was maintained above 80 wt% at all tested calcination temperatures. However, both isobutane conversion and isobutene selectivity decreased slightly with increasing calcination temperature. As a result, isobutene yield reduced from 61.8 wt% at 560 °C to 52.6 wt% at 800 °C. At the same time, both the selectivity to methane and n-butane were kept at a very low level, revealing that the sulfided 13Co/SiO2 catalyst was almost inactive for cracking and isomerization reactions. In summary, SiO2 seems to be the most appropriate support for Co-based catalysts in dehydrogenation. | ||
| Fig. 2 Dehydrogenation performance of sulfided Co-based catalysts: (a and b) 13Co/Al2O3; (c and d) 13Co/MgAl2O4; (e and f) 13Co/SiO2. | ||
Continuous activity test was further carried out to evaluate the stability of sulfided 13Co/SiO2 catalyst calcined at 560 °C, with the results shown in Fig. S1 in ESI.† Isobutane conversion decreased significantly with time on stream, especially at the initial stage, showing an obvious catalyst deactivation phenomenon. Fortunately, the decreased catalyst activity can be recovered by another sulfidation treatment. Within five continuous sulfidation-reaction cycles (see Fig. S2†), the initial isobutane conversion was maintained at a relatively steady level. To achieve continuous operation, a circulating fluidized bed unit equipped with a reactor for dehydrogenation, a regenerator for coke burning and a sulfidation section, is suggested to be employed in future industrial application.
Table 2 compares the adsorption amount of isobutane and isobutene (determined by pulse experiment) over different sulfided Co-based catalysts. The adsorption amount of isobutane decreased with increasing calcination temperature, which was consistent with the decrease of isobutane conversion. Combined with the results of activity test, it can be inferred that the adsorption of isobutane is not the bottleneck for isobutane dehydrogenation. As for isobutene, the adsorption amount decreased remarkably with increasing calcination temperature except for 13Co/SiO2 catalyst. And compared with SiO2-supported catalyst, the adsorption ability of 13Co/Al2O3 and 13Co/MgAl2O4 catalysts for isobutene was much stronger.
| Sample | Calcination temperature, °C | Adsorption amount, mL g−1 cat | |
|---|---|---|---|
| Isobutane | Isobutene | ||
| 13Co/Al2O3 | 560 | 0.68 | 2.47 |
| 800 | 0.49 | 1.42 | |
| 13Co/MgAl2O4 | 560 | 0.56 | 1.21 |
| 800 | 0.53 | 0.31 | |
| 13Co/SiO2 | 560 | 0.65 | 0.25 |
| 800 | 0.55 | 0.28 | |
The transient response signals of Ar and isobutene over different sulfided Co-based catalysts during the desorption procedure are illustrated in Fig. 3. In this figure, a larger signal delay extent between Ar and isobutene indicates a harder desorption behavior of the catalysts for isobutene and a higher possibility of secondary reactions. As for sulfided catalysts with different supports, the signal delay extent decreased in the following order: 13Co/Al2O3 > 13Co/MgAl2O4 > 13Co/SiO2, which well explained the worst selectivity to isobutene over Al2O3-supported catalysts. Moreover, with the increase of calcination temperature, the signal delay extent decreased obviously over Al2O3 and MgAl2O4-supported catalysts, which was consistent with the increase of selectivity to isobutene. Considering the above discussions, the desorption behaviors of the products are crucial to the selectivity.
 | ||
| Fig. 3 Transient response signals of Ar (dotted lines) and isobutene (solid lines) over different sulfided Co-based catalysts during the desorption procedure: (a) 13Co/Al2O3-560; (b) 13Co/Al2O3-800; (c) 13Co/MgAl2O4-560; (d) 13Co/MgAl2O4-800; (e) 13Co/SiO2-560; (f) 13Co/SiO2-800. | ||
 | ||
| Fig. 4 NH3-TPD profiles of different sulfided 13Co/support catalysts. | ||
With regard to sulfided Al2O3-supported catalysts, the acidity decreased with calcination temperature, which can possibly be attributed to the consumption of Al2O3 for the formation of CoAl2O4 on the catalyst (as evidenced by the XRD patterns in Fig. 1). And the acid amount of MgAl2O4-supported catalysts also exhibited the same variation trend with increasing calcination temperature.
Based on above discussions, it can be concluded that the decrease of acid amount results in an increasing selectivity to isobutene, which is consistent with other reports.41,44 That is, the presence of acid sites seems to have no promoting effect on the dehydrogenation performance. However, according to Abello et al.,43 acid sites on the catalyst facilitate the abstraction of the first hydrogen atom from C–H bond of alkane molecule, which is generally regarded as the rate-determining step. Some other researchers45,46 also believe that the presence of appropriate amount of acid sites facilitates the activation of alkanes. However, in this present study, the sulfided SiO2-supported catalysts calcined at different temperatures, almost with no acidity, exhibited a superior dehydrogenation activity and selectivity. This result indicates that the introduced sulfur also has the ability to capture the first hydrogen from isobutane molecule, and further initiates the dehydrogenation reaction. Therefore, it can be concluded that the acid sites are not necessary for this catalytic system.
According to above discussions, the support not only influences the interaction with active component but also its dispersion. As listed in Table 1, although the dispersion of Co species on SiO2 was the worst, the sulfided Co/SiO2 catalyst exhibited the highest TOF for isobutene formation. In contrast, alumina-supported catalysts with the best Co dispersion gave the lowest TOF value. This result suggests that Co dispersion is not the most important factor affecting the dehydrogenation performance in this catalytic system. In addition, it should also be noticed that the three evaluated supports with different acid properties, exhibited different adsorption–desorption behaviors for isobutane and isobutene and dehydrogenation performance as discussed above. From these aspects, silica, with no acidity, showed the highest prospect for application.
During catalyst preparation, the calcination temperature also influence the surface area,47–49 properties50,51 and phase composition52–55 of the catalyst remarkably. For all tested samples, the surface area decreased obviously with increasing calcination temperature, especially for silica-supported catalysts. Meanwhile, the acid amount also decreased for Al2O3 and MgAl2O4-supported catalysts, which was beneficial for obtaining a high selectivity to isobutene. In addition, the phase composition on the catalyst changed with calcination temperature. At high temperature, the interaction between Co species and the support became strong, and some solid state reactions took place. For instance, CoAl2O4 and Co2SiO4 formed on the surface of Co/Al2O3 and Co/SiO2 catalysts, respectively. The generation of CoAl2O4 decreased the catalyst acidity and inhibited secondary reactions effectively. In contrast, the formation of Co2SiO4 (difficult to be sulfided) probably led to the decrease of active sites, and thus worsened the dehydrogenation performance.
3.3 Mechanism of different promoters
 | ||
| Fig. 5 Catalytic performance of Sn or Cu promoted 13Co/SiO2 catalyst for isobutane dehydrogenation. | ||
To reduce the metal oxide on catalyst surface, the catalysts were prereduced by H2 for 2 h prior to reaction. During the activity test, the following three reactions (eqn (1)–(3)) probably take place over Co-based catalysts. It can be observed in Fig. 5 that, with hydrogen being supplied by deep dehydrogenation (eqn (1)) of isobutane,56 the reduced 13Co/SiO2 catalyst without any promoter exhibited extremely high activity for hydrogenolysis reaction and the selectivity to methane was up to 89.9 wt%.
| Deep dehydrogenation: i-C4H10 → 4C + 5H2 | (1) |
| Hydrogenolysis: i-C4H10 + 3H2 → 4CH4 | (2) |
| Dehydrogenation: i-C4H10 → i-C4H8 + H2 | (3) |
After introduction of Sn and Cu, the selectivity to isobutene increased sharply with increasing Sn and Cu contents, especially for Sn-modified catalysts. When 1 wt% Sn was introduced, the selectivity to isobutene increased from 0 to 54.6 wt%, and the selectivity to methane decreased to 5.2 wt%. Such results indicated that the introduction of Sn and Cu could suppress hydrogenlysis reaction effectively. However, isobutane conversion decreased obviously at the same time, making the modification less attractive.
To study the dehydrogenation performance of Sn and Cu promoted catalysts intuitively, pulse-mode isobutane dehydrogenation was carried out with the equipment of an online MS. Taking Cu-modified catalyst for example, when no Cu was introduced, the signals of isobutane (m/z = 58) and isobutene (m/z = 56) can hardly be detected except for the first pulse (as illustrated by the MS spectra in Fig. 6a). However, the signal of methane (m/z = 16) was obvious, demonstrating that 13Co/SiO2 catalyst exhibited extremely high hydrogenolysis activity, consistent with the results obtained by GC. After the introduction of Cu, the signal intensity of methane decreased gradually with increasing Cu content, meanwhile, that of isobutane increased obviously. In contrast, the signal intensity of isobutene increased for the initial introduction of Cu, and decreased subsequently. This observation can be attributed to the combined effects of decreasing isobutane conversion and increasing selectivity to isobutene.
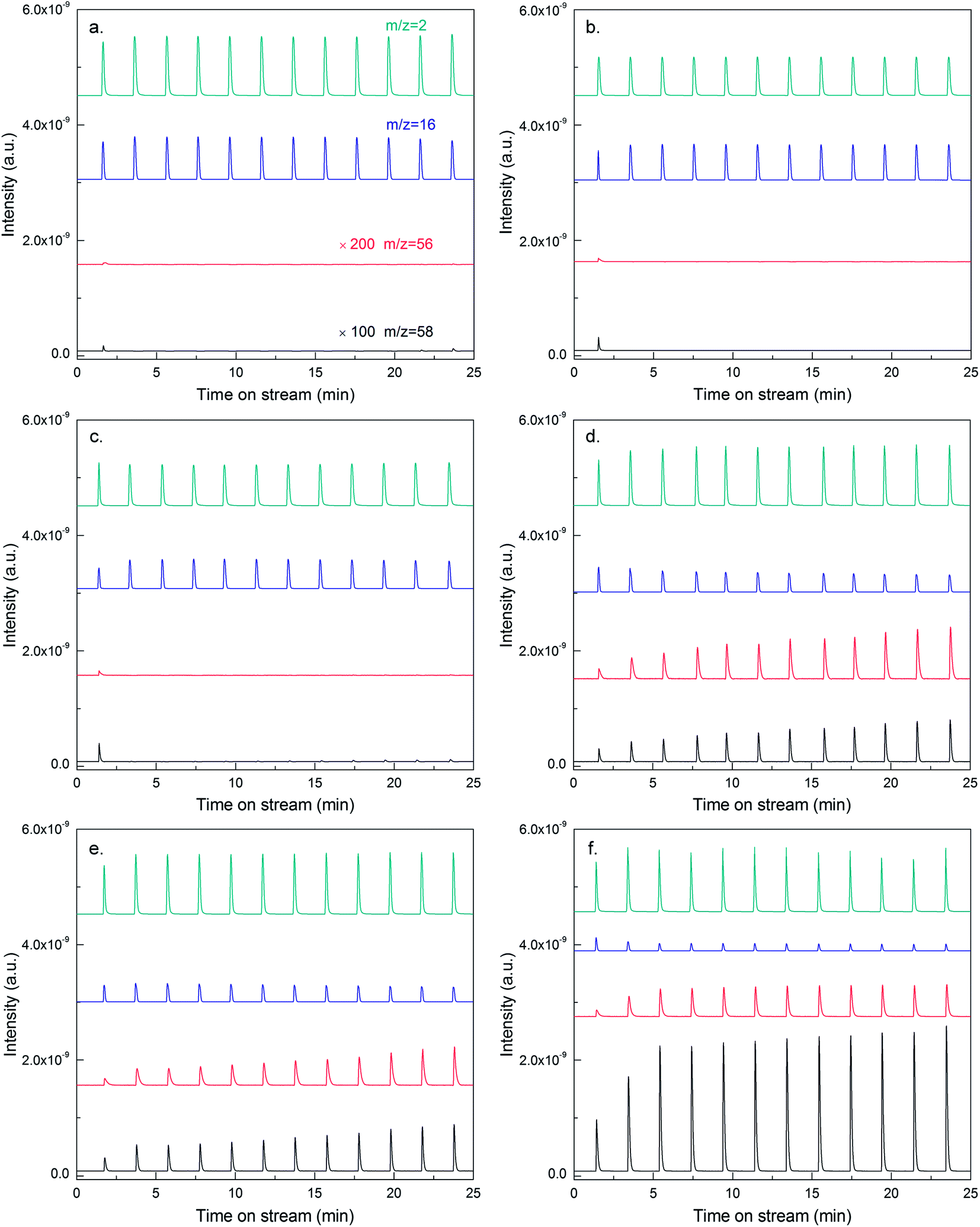 | ||
| Fig. 6 Mass spectra of pulse-mode isobutane dehydrogenation over Cu-modified Co/SiO2 catalysts: (a) 13Co/SiO2; (b) 1Cu–13Co/SiO2; (c) 2Cu–13Co/SiO2; (d) 3Cu–13Co/SiO2; (e) 4Cu–13Co/SiO2; (f) 5Cu–13Co/SiO2. | ||
As shown in Table 3, for 13Co/SiO2 catalysts modified by Sn and Cu, Co dispersion was much larger than that over the original 13Co/SiO2 catalyst (see Table 1), at the same time, the size of Co particles decreased obviously, indicating that the introduction of Sn or Cu destroyed the aggregated metallic species. In general, the performance of Sn or Cu promoted catalysts was similar to that of catalyst with low metal loading (i.e., high selectivity to isobutene but low activity). This fact revealed that the introduction of Sn or Cu destroyed metal ensembles through geometric or alloy effect, and suppressed hydrogenolysis reaction effectively. However, the dehydrogenation activity was not improved, and isobutene yield only reached to 27.5 wt% and 14.1 wt% over Sn and Cu promoted catalysts, respectively, much lower than S-modified catalyst. And the reason might lie in the difference of binding affinity between S and Sn or Cu to hydrogen atom in isobutane molecule.
| Sample | SBETa, m2 g−1 | Cosurf. density, atomCo/nm2 | Co dispersionb, % | DCoc, nm |
|---|---|---|---|---|
| a Specific surface area.b Co dispersion was determined by H2 chemisorption, referred to the molar ratio of surface Co to bulk Co, with the latter being calculated from the cobalt nitrate impregnation.c Particle size of Co was determined by H2 chemisorption.d Contents of Cu and Sn in the catalyst were 5 wt%.e Content of Cl in the catalyst was 3.7 wt%, which was determined by XRF. | ||||
| 13Co/SiO2-Snd | 221 | 4.4 | 34.1 | 2.5 |
| 13Co/SiO2-Cud | 218 | 4.5 | 31.9 | 2.7 |
| 13Co/SiO2-Cle | 230 | 4.2 | 27.1 | 3.2 |
Based on our previous studies,3,7,10 involving combined characterizations of H2-TPR, SEM, XPS and purposive activity tests, the catalytic cycle of cobalt sulfide catalyst for isobutane dehydrogenation can be speculated as Scheme 1. An isobutane molecule adsorbs on the surface of the catalyst dissociatively, forming an intermediate with cobalt bonded to the tertiary butyl and sulfur bonded to the left hydrogen. Subsequently, the bond between β-hydrogen and sulfur weakens the C–H bond in isobutane, leading to the formation of a molecule of hydrogen. The left intermediate with a π-bonded alkene releases an isobutene molecule, meanwhile the active center is recovered. During this procedure, the high binding affinity of sulfur to two hydrogen atoms facilitates the formation of hydrogen, and the reaction equilibrium is shifted towards the right side. As a result, the dehydrogenation activity is improved dramatically after sulfur modification. However, as for Sn and Cu, the binding ability to hydrogen is not strong enough, therefore, the effect of Sn and Cu introduction only involves breaking metal ensembles and inhibiting hydrogenolysis reaction.
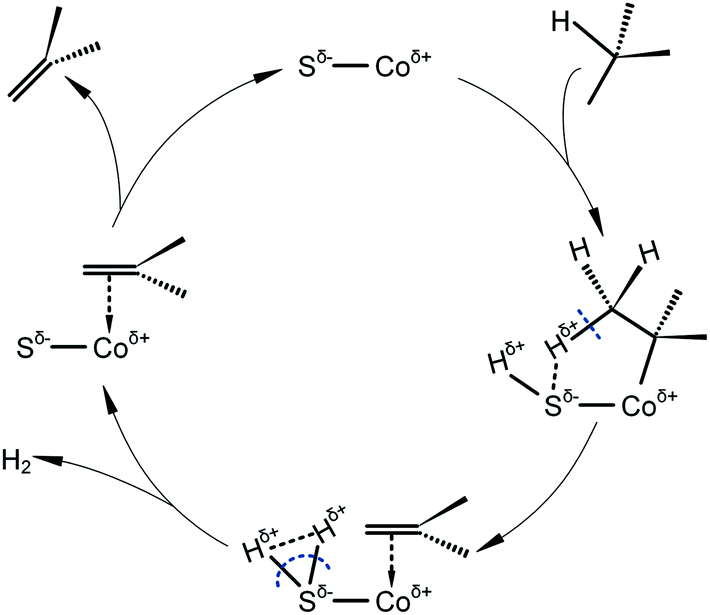 | ||
| Scheme 1 Proposed catalytic cycle of sulfided Co/SiO2 catalyst for isobutane dehydrogenation. | ||
Before the activity test, the catalyst sample was prereduced by hydrogen for 2 h, and then the reaction lasted for 2 h. To determine the content of chlorine on 13Co/SiO2–Cl catalyst before and after reaction, elemental analysis was further carried out by XRF. Cl content of the fresh catalyst was 3.72 wt%, indicating that part of Cl was removed from the catalyst during the calcination process. After 2 h reaction, Cl content on the catalyst was still up to 3.58 wt%, remaining almost stable. It can be seen in Fig. 7 that, compared with 13Co/SiO2 catalyst, the selectivity to isobutene increased remarkably after chlorine introduction, meanwhile, the selectivity to methane decreased sharply, indicating that the hydrogenolysis reaction was effectively inhibited. The decreasing catalyst activity with time on stream was probably resulted from the coke formation. Meanwhile, under the coverage and isolation effects of coke, the selectivity to isobutene increased consequently. However, isobutane conversion was constantly lower than 10 wt% for the continuous 2 h reaction. This result indicates that, although chlorine has the ability to bond with hydrogen, the introduction of chlorine could not improve the dehydrogenation activity as well. Primarily, it is worth mentioning that one chlorine atom can only bind to one hydrogen atom and can not capture β-hydrogen atom at the same time. Moreover, the distance between two chlorine atoms on the catalyst surface is probably larger than that between the first hydrogen and β-hydrogen of one isobutane molecule. And even if two chlorine atoms capture the two hydrogen atoms of one isobutane simultaneously, the formation of hydrogen would still be restricted due to the untouchable distance. Therefore, the dehydrogenation activity is limited after chlorine modification.
 | ||
| Fig. 7 Catalytic performance of Cl promoted 13Co/SiO2 catalyst for isobutane dehydrogenation. | ||
Based on the above discussions, the promoting mechanisms of different additives are summarized and depicted in Scheme 2. In general, an efficient promoter should at least satisfy the following requirements: the first is to effectively suppress the formation of metal ensembles, which are active for hydrogenolysis reaction; the second is a strong bonding affinity to hydrogen atoms; lastly, to shift the reaction equilibrium towards right side, the promoter should have the ability to combine with two hydrogen atoms simultaneously and facilitate the formation of hydrogen.
 | ||
| Scheme 2 Schematic of promoting mechanism of different methods. | ||
4. Conclusions
Based on the active phase of metal sulfide catalysts responsible for dehydrogenation and how they interact with isobutane molecules explored in our pervious studies, the effects of support, calcination temperature and some other promoters rather than sulfur on dehydrogenation performance of Co-based catalyst have been further investigated to optimize this novel class of catalyst in this paper.Experimental results indicate that, both the support and calcination temperature affect Co dispersion, catalyst acid properties and the interaction between support and Co species significantly, and further influence the dehydrogenation performance. However, Co dispersion does not seem to be an important factor in this catalytic system, and the acid sites adsorb isobutene strongly and further facilitate undesired secondary reactions. As a result, silica-supported catalysts, with no acidity, exhibited the best dehydrogenation performance. But the formation of Co2SiO4 (hardly to be sulfided) over Co/SiO2 at high calcination temperatures led to an decrease of active sites and thus worsened the dehydrogenation performance. In contrast, high calcination temperature was beneficial for obtaining a high selectivity to isobutene for Al2O3 and MgAl2O4-supported catalysts.
Furthermore, detailed investigations into the effects of different additives indicate that, an efficient promoter should at least possess the following features: inhibiting the formation of metal ensembles to decrease hydrogenolysis reaction, and a strong binding affinity to two hydrogen atoms of one additive atom for enhancing dehydrogenation activity.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. U1362201) and the National 973 Program of China (no. 2012CB215006).References
- G. Wang, C. Li, H. Shan and W. Wu, Ind. Eng. Chem. Res., 2013, 52, 13297–13304 CrossRef CAS.
- D. E. Resasco, B. K. Marcus, C. S. Huang and V. A. Durante, J. Catal., 1994, 146, 40–55 CrossRef CAS.
- G. Wang, C. Gao, X. Zhu, Y. Sun, C. Li and H. Shan, ChemCatChem, 2014, 6, 2305–2314 CAS.
- R. W. Coughlin, A. Hasan and K. Kawakami, J. Catal., 1984, 88, 163–176 CrossRef CAS.
- P.-C. Liao, T. H. Fleisch and E. E. Wolf, J. Catal., 1982, 75, 396–403 CrossRef CAS.
- M. Luo, Q. Wang, G. Li, X. Zhang, L. Wang and L. Han, Catal. Commun., 2013, 35, 6–10 CrossRef CAS PubMed.
- G. Wang, Z. Meng, J. Liu, C. Li and H. Shan, ACS Catal., 2013, 3, 2992–3001 CrossRef CAS.
- G. Wang, N. Sun, C. Gao, X. Zhu, Y. Sun, C. Li and H. Shan, Appl. Catal., A, 2014, 478, 71–80 CrossRef CAS PubMed.
- Y.-n. Sun, L. Tao, T. You, C. Li and H. Shan, Chem. Eng. J., 2014, 244, 145–151 CrossRef CAS PubMed.
- G. Wang, C. Li and H. Shan, ACS Catal., 2014, 1139–1143 CrossRef.
- M. Chen, J. L. Wu, Y. M. Liu, Y. Cao, L. Guo, H. Y. He and K. N. Fan, Appl. Catal., A, 2011, 407, 20–28 CrossRef CAS PubMed.
- R. Grabowski, Catal. Rev.: Sci. Eng., 2006, 48, 199–268 CAS.
- M. Bhasin, J. McCain, B. Vora, T. Imai and P. Pujado, Appl. Catal., A, 2001, 221, 397–419 CrossRef CAS.
- F. Hoxha, B. Schimmoeller, Z. Cakl, A. Urakawa, T. Mallat, S. E. Pratsinis and A. Baiker, J. Catal., 2010, 271, 115–124 CrossRef CAS PubMed.
- E. Schmidt, F. Hoxha, T. Mallat and A. Baiker, J. Catal., 2010, 274, 117–120 CrossRef CAS PubMed.
- C. Chen, J. Cao, M. Cargnello, P. Fornasiero and R. J. Gorte, J. Catal., 2013, 306, 109–115 CrossRef CAS PubMed.
- A. M. Abdel-Mageed, S. Eckle, H. G. Anfang and R. J. Behm, J. Catal., 2013, 298, 148–160 CrossRef CAS PubMed.
- K. Lv, Q. Xiang and J. Yu, Appl. Catal., B, 2011, 104, 275–281 CrossRef CAS PubMed.
- L. Ma, J. Li, H. Arandiyan, W. Shi, C. Liu and L. Fu, Catal. Today, 2012, 184, 145–152 CrossRef CAS PubMed.
- Z. Boukha, L. Fitian, M. López-Haro, M. Mora, J. R. Ruiz, C. Jiménez-Sanchidrián, G. Blanco, J. J. Calvino, G. A. Cifredo and S. Trasobares, J. Catal., 2010, 272, 121–130 CrossRef CAS PubMed.
- D. Duvenhage and N. Coville, Appl. Catal., A, 2002, 233, 63–75 CrossRef CAS.
- V. Ravat, I. Nongwe, R. Meijboom, G. Bepete and N. J. Coville, J. Catal., 2013, 305, 36–45 CrossRef CAS PubMed.
- F. B. Passos, M. Schmal and M. A. Vannice, J. Catal., 1996, 160, 106–117 CrossRef CAS.
- F. B. Passos, D. A. G. Aranda and M. Schmal, J. Catal., 1998, 178, 478–488 CrossRef CAS.
- J. Z. Zhang, Y.-T. Tsai, K. L. Sangkaewwattana and J. G. Goodwin Jr, J. Catal., 2011, 280, 89–95 CrossRef CAS PubMed.
- G. S. Ranhotra, A. T. Bell and J. A. Reimer, J. Catal., 1987, 108, 40–49 CrossRef CAS.
- R. J. Lobo-Lapidus and B. C. Gates, J. Catal., 2009, 268, 89–99 CrossRef CAS PubMed.
- C. R. Apesteguía and J. Barbier, J. Catal., 1982, 78, 352–359 CrossRef.
- J. C. Serrano-Ruiz, A. Sepúlveda-Escribano and F. Rodríguez-Reinoso, J. Catal., 2007, 246, 158–165 CrossRef CAS PubMed.
- R. D. Cortright and J. A. Dumesic, J. Catal., 1994, 148, 771–778 CrossRef CAS.
- M. C. Román-Martínez, J. A. Maciá-Agulló, I. M. J. Vilella, D. Cazorla-Amorós and H. Yamashita, J. Phys. Chem. C, 2007, 111, 4710–4716 Search PubMed.
- X. Wang, L. Altmann, J. Stöver, V. Zielasek, M. Bäumer, K. Al-Shamery, H. Borchert, J. Parisi and J. Kolny-Olesiak, Chem. Mater., 2012, 25, 1400–1407 CrossRef.
- J. H. Sinfelt, J. Carter and D. Yates, J. Catal., 1972, 24, 283–296 CrossRef CAS.
- G. A. Martin and J. A. Dalmon, J. Catal., 1982, 75, 233–242 CrossRef CAS.
- K. Lu and B. J. Tatarchuk, J. Catal., 1987, 106, 166–175 CrossRef CAS.
- K. Lu and B. J. Tatarchuk, J. Catal., 1987, 106, 176–187 CrossRef CAS.
- E. T. Iyagba, T. Eddy Hoost, J. U. Nwalor and J. G. Goodwin Jr, J. Catal., 1990, 123, 1–11 CrossRef CAS.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, ACS Catal., 2013, 4, 182–186 CrossRef.
- R. Grabowski, B. Grzybowska, K. Samson, J. Słoczyński, J. Stoch and K. Wcisło, Appl. Catal., A, 1995, 125, 129–144 CrossRef CAS.
- K. Chen, S. Xie, E. Iglesia and A. T. Bell, J. Catal., 2000, 189, 421–430 CrossRef CAS.
- L. Madeira, M. Portela, C. Mazzocchia, A. Kaddouri and R. Anouchinsky, Catal. Today, 1998, 40, 229–243 CrossRef CAS.
- S. Ge, C. Liu and L. Wang, Chem. Eng. J., 2001, 84, 497–502 CrossRef CAS.
- M. Abello, M. Gomez and O. Ferretti, Appl. Catal., A, 2001, 207, 421–431 CrossRef CAS.
- K. Chen, A. T. Bell and E. Iglesia, J. Phys. Chem. B, 2000, 104, 1292–1299 CrossRef CAS.
- J. C. Vedrine, J. M. M. Millet and J.-C. Volta, Catal. Today, 1996, 32, 115–123 CrossRef CAS.
- A. Pantazidis, A. Auroux, J. M. Herrmann and C. Mirodatos, Catal. Today, 1996, 32, 81–88 CrossRef CAS.
- V. R. Choudhary, D. K. Dumbre, N. S. Patil, B. S. Uphade and S. K. Bhargava, J. Catal., 2013, 300, 217–224 CrossRef CAS PubMed.
- E. Aneggi, N. J. Divins, C. de Leitenburg, J. Llorca and A. Trovarelli, J. Catal., 2014, 312, 191–194 CrossRef CAS PubMed.
- C. Kang, L. Jing, T. Guo, H. Cui, J. Zhou and H. Fu, J. Phys. Chem. C, 2008, 113, 1006–1013 Search PubMed.
- H. Yamashita, Y. Horiuchi, S. Imaoka, S. Nishio, N. Nishiyama and K. Mori, Catal. Today, 2008, 132, 146–152 CrossRef CAS PubMed.
- P. Górska, A. Zaleska, E. Kowalska, T. Klimczuk, J. W. Sobczak, E. Skwarek, W. Janusz and J. Hupka, Appl. Catal., B, 2008, 84, 440–447 CrossRef PubMed.
- F. Liu, K. Asakura, H. He, Y. Liu, W. Shan, X. Shi and C. Zhang, Catal. Today, 2011, 164, 520–527 CrossRef CAS PubMed.
- D. Y. Yoon, J.-H. Park, H.-C. Kang, P. S. Kim, I.-S. Nam, G. K. Yeo, J. K. Kil and M.-S. Cha, Appl. Catal., B, 2011, 101, 275–282 CrossRef CAS PubMed.
- K. Li, H. Wang, Y. Wei and D. Yan, Int. J. Hydrogen Energy, 2011, 36, 3471–3482 CrossRef CAS PubMed.
- X. Guo, D. Mao, G. Lu, S. Wang and G. Wu, Catal. Commun., 2011, 12, 1095–1098 CrossRef CAS PubMed.
- E. A. Redekop, V. V. Galvita, H. Poelman, V. Bliznuk, C. Detavernier and G. B. Marin, ACS Catal., 2014, 4, 1812–1824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08849b |
| This journal is © The Royal Society of Chemistry 2014 |