
Fe(ii)(aq) uptake of Mg(ii)–Al(iii)/Fe(iii)–SO4/CO3 HTLCs under alkaline conditions: adsorption and solid state transformation mechanisms†
Abstract
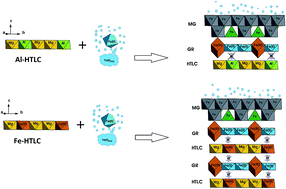
Abiotic reduction of Mg(II)–Al(III)/Fe(III)–SO4/CO3 hydrotalcites (HTLCs) was investigated under three anoxic abiotic reaction conditions: (1) a target pH of 8 and 10 mM Fe(II)(aq), (2) a target pH of 8 and 0.5 mM Fe(II)(aq), and (3) a target pH of 10 and 0.5 mM Fe(II)(aq). Under both Fe(II)(aq) concentrations and at the targeted pH of 8, the greatest Fe(II)(aq) uptake, Mg(II)(aq) release, and least deviation in pH was observed for Fe-based HTLCs (Mg(II)–Fe(III)–SO4/CO3) irrespective of the interlayer ion (SO4 or CO3). In contrast, the Al-based HTLCs showed the greatest pH drift from the target pH of 8; this phenomenon was attributed to the remaining total Fe(II + III) in solution. At the target pH of 10 and 0.5 mM Fe(II)(aq), the pH of the Al-based HTLCs was greater than the Fe-based HTLCs despite 99% of the Fe(II) being removed from solution in both cases. The greatest Mg(II)(aq) release was again observed for the Fe-based HTLCs. Unique to these reaction conditions was the readsorption of the released Mg(II)(aq) by the Mg–Fe–CO3 HTLC. The major reaction phases produced from the adsorption of Fe(II)(aq) onto Mg(II)–Al(III)/Fe(III)–SO4/CO3 HTLCs were green rust (GR) and magnetite (MG). Two types of phase transformation mechanisms are suggested to occur during the adsorption of Fe(II)(aq) onto Mg(II)–Al(III)/Fe(III)–SO4/CO3 HTLCs, both dependent upon the metal ion (Al vs. Fe) and independent of the interlayer ions (SO4 or CO3). For Fe-based HTLCs (Mg–Fe–SO4/CO3), surface adsorption of Fe(II)(aq) and surface precipitation allows electron transfer (between Fe(II) ↔ Fe(III)) and subsequent atom exchange to occur at the surface as well as within the bulk HTLC structure. For the Al-based HTLCs (Mg–Al–SO4/CO3), this surface adsorption of Fe(II)(aq) and surface precipitation still occurs but electron transfer is limited to the precipitated surface phase due to the redox inactivity of the Al(III) groups in the HTLC structure.
 Please wait while we load your content...
Please wait while we load your content...

