
Facile and controllable synthesis of carbon-encapsulating carbonate apatite nanowires from biomass containing calcium compounds such as CaC2O4 and CaCO3†
Abstract
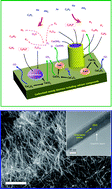
We report the synthesis of carbon-encapsulating carbonate apatite nanowires through vapor–solid growth by heat-treatment of biomass comprising calcium compounds such as CaC2O4 or CaCO3 at 900 °C using both PH3 and C2H2 as the reactants. The thermal decomposition of CaC2O4 or CaCO3 to CaO with increasing temperature (CaC2O4 → CaCO3 + CO → CaO + CO2) is the key to achieving the growth of such core–shell nanowires. First, vapor-phase reactions between the gaseous calcium species generated from the derived CaO and gaseous molecules derived from thermal reactions of the reactants (PH3 and C2H2) lead to the oriented growth of core–shell nanowires along the [001] plane. Second, the CO2 generated during the decomposition of CaCO3 may be primarily responsible for the incorporation of carbonate ions into the apatite structure. Nanowire growth with knots along the growth direction reveals that our approach is very controllable. Additional demonstrations using kenaf fibers further verify that other types of biomass too are usable.
 Please wait while we load your content...
Please wait while we load your content...

