
Fabrication of a novel magnetic yolk–shell Fe3O4@mTiO2@mSiO2 nanocomposite for selective enrichment of endogenous phosphopeptides from a complex sample†
Abstract
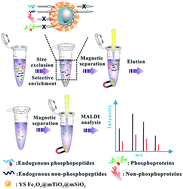
A yolk–shell nanocomposite composed of a magnetic mesoporous anatase TiO2 (Fe3O4@mTiO2) core, a medium cavity and an outermost mesoporous silica (mSiO2) shell was successfully fabricated. Due to a combination of the strong magnetic response, improved diffusion of peptides, numerous affinity sites towards phosphopeptides and the size-exclusion effect, the nanocomposite demonstrated high enrichment efficacy and selectivity towards endogenous phosphopeptides from human serum.
 Please wait while we load your content...
Please wait while we load your content...

