
K2S2O8/arenesulfinate: an unprecedented thiolating system enabling selective sulfenylation of indoles under metal-free conditions†
Honghua Rao *a,
Ping Wang‡
b,
Jianchun Wang‡a,
Zhongfeng Li‡a,
Xinzhan Suna and
Shengli Caoa
*a,
Ping Wang‡
b,
Jianchun Wang‡a,
Zhongfeng Li‡a,
Xinzhan Suna and
Shengli Caoa
aDepartment of Chemistry, Capital Normal University, Beijing 100048, P. R. China. E-mail: honghua.rao@gmail.com; Fax: +86-10-68902493
bSchool of Chemical Engineering, Beijing Institute of Petrochemical Technology, Beijing 102617, P. R. China
First published on 26th September 2014
Abstract
An unprecedented thiolating system K2S2O8/arenesulfinate is described for selective sulfenylation of indoles in CH3CN/H2O. This metal-free strategy enables a simple, efficient and environment-benign approach to the pharmaceutically important candidates, 3-arylthioindoles. Catalytically reactive halogen and aryl groups are well tolerated.
The indole scaffold is a prominent and privileged functionality occurring in many biologically important compounds,1 and consequently, it has been attracting considerable attention from synthetic chemists to develop strategies for the construction and chemical modification of the indole ring.2 Among these strategies, the sulfenylation of indoles is particularly attractive owing to the promising therapeutic applications of the resulting thiolated indoles, in which the 3-sulfanylindoles could serve as potential candidates for the treatment of cancer,3 HIV,4 allergies,5 and heart disease (Fig. 1).6
 | ||
| Fig. 1 Examples of thiolated indoles with biological activities. | ||
Conventional methods for the syntheses of 3-sulfanylindoles rely on the transition-metal-catalyzed cross-couplings between 3-(pseudo)halogenated indoles and thiols.7 Given the electron-rich nature of indole rings, direct C–H sulfenylation of indoles with electrophiles has aroused much more interests in recent years due to their advantages from both step- and atom-economy points of view in industrial and green chemistry. In this regard, a variety of thiolating agents such as thiols, disulfides and activated sulphur reagents were employed with the C–H sulfenylation occurring smoothly catalyzed by vanadium,8 magnesium,9 iron,10 cerium,11 copper,12 palladium13 and ruthenium catalysts.14 Some intriguing achievements via direct C–H sulfenylations to 3-thioindoles were further developed under metal-free conditions (Scheme 1a).15 For instance, disulfides as the thiolating agents promoted by N-bromosuccinimides,16 carbonates,17 persulfate18 and I219 displayed efficient reactivities in the absence of any transition metals. Very recently, sulfonyl hyrazide has been proved to be a successful sulfenylation agent with catalytic amount of I2 molecule (Scheme 1b).20 Besides, Deng21 and other groups22 found that arenesulfinates can also undergo sulfenylation reactions with indoles using molecule I2 as the organocatalyst similarly (Scheme 1c). But it should be noted that DMSO and phosphite or phosphine are crucial for the deoxygenation of arylsulfonyl to arylthio group when using arenesulfinate as the thiolating agent, and thus might give off unpleasant odors (for dimethyl sulfide was generated from DMSO). Therefore, it is still desirable to explore cheaper, simpler and more environment-friendly sulfenylation systems for the syntheses of 3-sulfanylindoles. It is well known that K2S2O8 can play as a radical initiator to generate sulfonyl radicals from sulfinates,23 however, to the best of our knowledge, no examples were given that K2S2O8 acted as promoters for deoxygenation reactions. Herein, we disclose an unprecedented thiolating system, K2S2O8/arenesulfinate, for the direct selective sulfenylation of indoles, affording 3-arylthioindoles in moderate to excellent yields in CH3CN/H2O (v/v ratio 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) without any other additives (Scheme 1d).
:1) without any other additives (Scheme 1d).
 | ||
| Scheme 1 C3-sulfenylation of indoles with various thiolating systems. | ||
Since ammonium halide/TBHP organocatalytic system has already been proved to be efficient for C–H functionalizations by our group,24 the direct C–H sulfenylation of indole (1.0 equiv.) was commenced by testing different ammonium halides at 100 °C under inert atmosphere, with TBHP as the oxidant, p-tolylsulfinate (1.5 equiv.) as the sulfenylation (or sulfonylation) agent, and CH3CN (1.0 mL) as the solvent. As screened in Table 1, either TBAB/TBHP or TBAI/TBHP as the organocatalytic system afforded nearly full recovery of indole (Table 1, entries 1–3), while the unexpected product 3a, 3-p-tolylthioindole (Fig. 2), was obtained in 20% isolated yield if replacing TBHP with K2S2O8 (2.0 equiv.) (entry 4). Inspiringly, the reaction efficiency increased to 46% when only subjecting K2S2O8 (1.0 equiv.) to the reaction system (entries 5 and 6).
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | [Ox] (equiv.) | Solvent | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: indole (0.2 mmol), p-tolylsulfinate (0.3 mmol, 1.5 equiv.), solvent (1.0 mL), reaction time (24 h), under N2.b Isolated yield.c CH3CN/H2O 10:1 (v/v ratio).d Under air.e CH3CN/H2O 1:1 (v/v ratio).f p-Tolylsulfinate (0.4 mmol, 2.0 equiv.).g p-Tolylsulfinate (0.1 mmol, 0.5 equiv.). Cat. = catalyst, [Ox] = oxidant, Temp. = temperature, TBAB = tetra-n-butylammonium bromide, TBAI = tetra-n-butylammonium iodide, TBHPdec = tert-butyl hydroperoxide (5.0–6.0 M in decane), TBHPaq = tert-butyl hydroperoxide (70% aqueous solution). |
|||||
| 1 | TBAB (10) | TBHPdec (2.0) | CH3CN | 100 | 0 |
| 2 | TBAI (10) | TBHPdec (2.0) | CH3CN | 100 | Trace |
| 3 | TBAI (10) | TBHPaq (2.0) | CH3CN | 100 | <10 |
| 4 | TBAI (10) | K2S2O8 (2.0) | CH3CN | 100 | 20 |
| 5 | — | K2S2O8 (2.0) | CH3CN | 100 | 32 |
| 6 | — | K2S2O8 (1.0) | CH3CN | 100 | 46 |
| 7 | — | (NH4)2S2O8 (1.0) | CH3CN | 100 | 37 |
| 8 | — | Na2S2O8 (1.0) | CH3CN | 100 | 41 |
| 9 | — | Oxone (1.0) | CH3CN | 100 | 41 |
| 10 | — | K2S2O8 (1.0) | EtOAc | 100 | <10 |
| 11 | — | K2S2O8 (1.0) | DMF | 100 | 35 |
| 12 | — | K2S2O8 (1.0) | p-Dioxane | 100 | 0 |
| 13c | — | K2S2O8 (1.0) | CH3CN/H2O | 100 | 89 |
| 14c | — | K2S2O8 (1.0) | CH3CN/H2O | 120 | 81 |
| 15c | — | K2S2O8 (1.0) | CH3CN/H2O | 80 | 74 |
| 16c,d | — | K2S2O8 (1.0) | CH3CN/H2O | 100 | 70 |
| 17e | — | K2S2O8 (1.0) | CH3CN/H2O | 100 | 71 |
| 18c | — | K2S2O8 (0.5) | CH3CN/H2O | 100 | 65 |
| 19c | — | K2S2O8 (1.0) | CH3CN/H2O | 100 | 88f, 76g |

 | ||
| Fig. 2 Structure of compound 3a determined by X-ray diffraction. | ||
Since persulfate could greatly improve the direct C–H sulfenylation of indole, several persulfates such as (NH4)2S2O8, Na2S2O8 and oxone were tested and particularly K2S2O8 gave the highest (46%) yield of product 3a (entries 6–9). In the meanwhile, the efficiency of the direct sulfenylation shows a strong dependence on solvents. For example, a sharp decrease was observed when using EtOAc or DMF as the solvent (entries 10 and 11). Moreover, even no sulfenylation product was observed when conducting the reaction in p-dioxane (entry 12). Fortunately, a small amount of water (v/v ratio to CH3CN 1:10) enhanced the reaction efficiency dramatically, giving 3a in the highest yield of 89% among all the solvents examined (entry 13). Other endeavours to improve the yield of 3a were attempted, such as altering the reaction temperature, reducing the loading of K2S2O8, running the reaction in air atmosphere or loading different amounts of reagent p-tolylsulfinate, but none of them could lead to higher sulfenylation reactivity (entries 14–19).
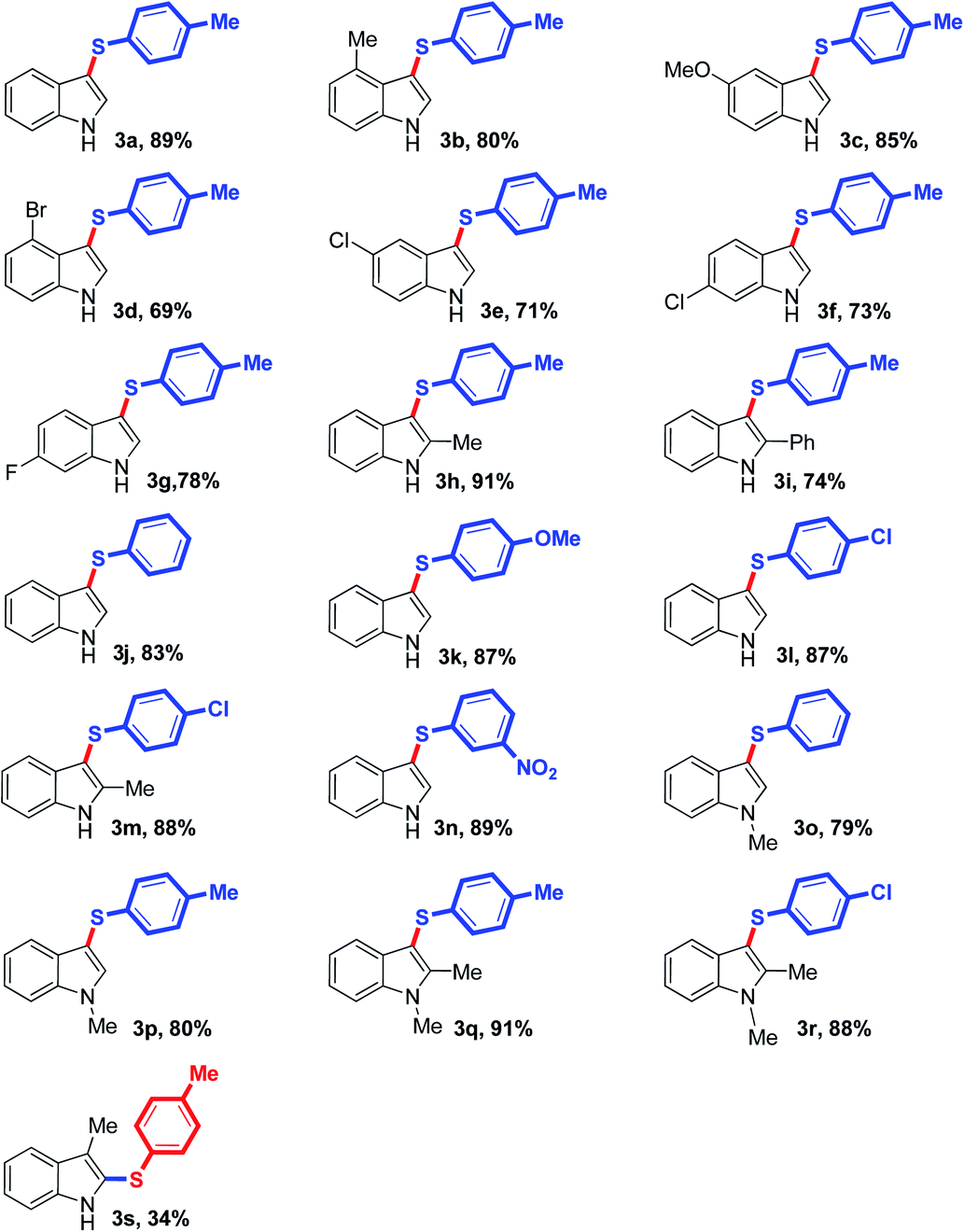
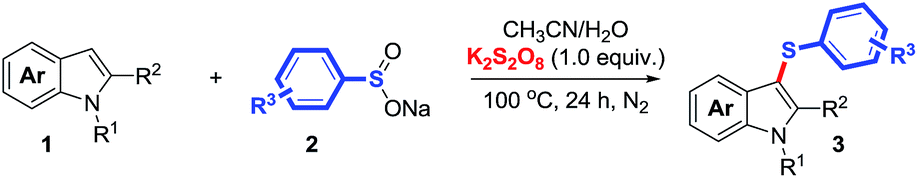
With the optimized reaction conditions in hand, the substrate scope was explored at 100 °C for 24 h under N2 atmosphere, using K2S2O8/arenesulfinate as the thiolating system and CH3CN/H2O (v/v ratio 10:1) as the solvent. As summarized in Table 2, free NH-indoles that might exhibit site-selectivity challenges were reacted with p-tolylsulfinate to the corresponding 3-arylthioindoles exclusively in good to excellent yields (Table 2, cf. 3a–g). Indoles with electron-donating substituents such as methyl and methoxyl groups (cf. 3b–c) exhibited higher reactivities over that with electron-withdrawing groups such as fluoro, chloro and bromo groups (cf. 3d–g). To some extent, the sulfenylation efficiency is affected by steric hindrance, for example, 4-methylindole as the substrate afforded slightly lower yield than indole did (cf. 3a–b). Other arylsulfinates bearing electron-donating group (e.g. methoxy group) or electron-withdrawing groups (e.g. chloro and nitro group) were also tested, and all of them reacted smoothly with indoles, yielding the desired 3-arylthioindoles highly efficiently (cf. 3j–n). Besides, N-protected indoles could couple with arenesulfinates effectively as well (cf. 3o–r). The availability of 3-substituted indoles for this sulfenylation strategy was also explored, and the desired 3-methyl-2-(p-tolylthio)indole product from 3-methylindole was obtained, albeit in a much lower yield (cf. 3s).25 Particularly, it is noteworthy that the tolerance of catalytically reactive substituents such as halides and phenyl groups enables further chemical modifications of the desired 3-arylthioindoles (cf. 3d–g, 3i, 3k–n and 3r), and thus promises the discovery of various 3-sulfanylindoles with important therapeutic value. To investigate the practical application of this transformation in organic synthesis, we conducted gram-scale synthesis of 3a. The desired product was produced without any significant decrease in efficiency (86% versus 89% for the reaction on a 0.2 mmol scale for 3a; Scheme 2).
|
|---|
| a Reaction conditions: indole (0.2 mmol), arenesulfinate (0.3 mmol, 1.5 equiv.), K2S2O8 (0.2 mmol, 1.0 equiv.), CH3CN/H2O (1.0 mL, v/v ratio 10:1), reaction temperature (100 °C), reaction time (24 h), under N2. Isolated yields were given unless otherwise noted. |
 |
 | ||
| Scheme 2 Gram-scale synthesis of 3a. | ||
To gain some insights into the sulfenylation pathway with this unprecedented thiolating system, some control experiments were conducted under various reaction conditions. As S2O82− usually serves as a radical initiator at elevated temperature,26 the radical process is a preferred consideration. Therefore, a radical-trapping experiment was carried out by introducing TEMPO into the standard conditions (Scheme 3a). And indeed, the desired sulfenylation reaction did not occur, thus indicating that this transformation is likely to involve a radical intermediate. Meanwhile, the reaction also did not take place in the absence of K2S2O8 (Scheme 3b), whereas it afforded only 52% of the desired product 3a and 29% of the undesired product 2-p-tosylindole 4 when employing only 1.0 equivalent of p-tolylsulfinate (Scheme 3c). These observations suggest that both K2S2O8 and excess amount of arenesulfinates are necessary for the radical sulfenylation reaction, and most importantly, arylsulfonyl radical is involved in this process. Furthermore, deoxygenation of 3-p-tosylindole (or 2-p-tosylindole) with K2S2O8 (1.0 equiv.) in CH3CN/H2O (v/v ratio 10:1) or under standard reaction conditions was conducted to explore that whether 3-arylsulfonylindole (or 2-arylsulfonylindole) is the key intermediate for this sulfenylation strategy, but unfortunately, full recovery of the starting material was observed (Scheme 3d). This result indicates that the tosylated indole is unlikely to serve as the key intermediate in the sulfenylation pathway, and arylsulfonyl radical most probably undergoes deoxygenation to arylthio radical (or cation) before finally introduced to indole ring.
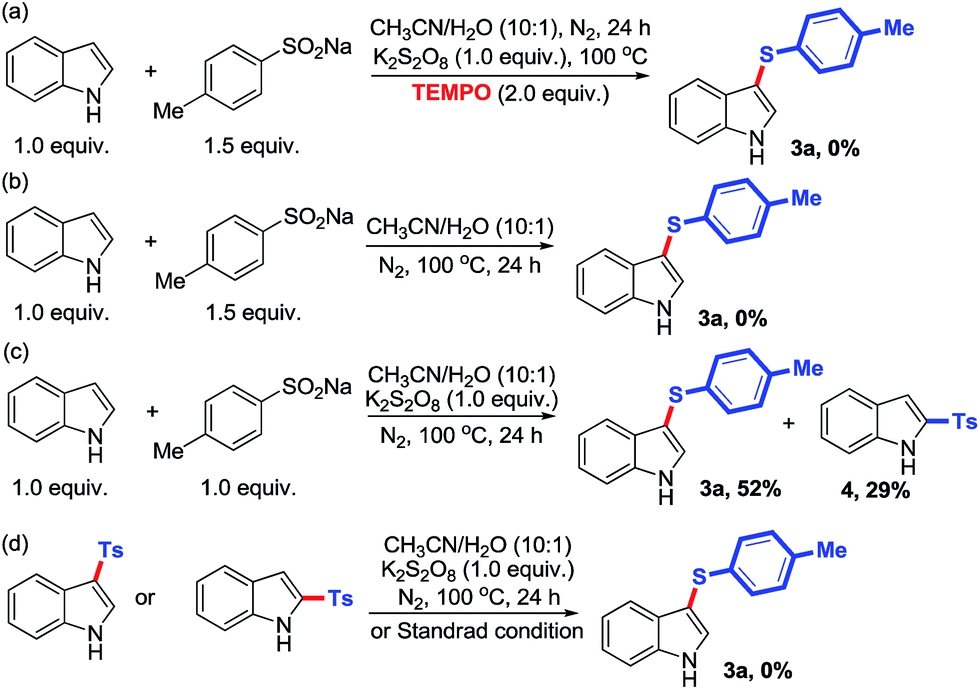 | ||
| Scheme 3 Control experiments. | ||
Based on previous work23,27 and the above results, a tentative mechanism for this unprecedented sulfenylation reaction is proposed in Scheme 4. The reaction is initiated via the homolysis of K2S2O8 to radical anion SO4˙− (Scheme 4a), which could promote the formation of arylsulfonyl radical A upon reaction with arenesulfinate (Scheme 4b), and the resulting radical A undergoes deoxygenation with excess amount of arylsulfinate and SO42− to afford arylthio radical B (Scheme 4c). The radical addition of B to indole ring gives intermediate C, which is believed to undergo the direct deprotonation to release the final product 3 (Scheme 4d). (However, multiple pathways may be involved in this transformation. A thorough mechanistic study is needed to unravel the mechanistic intricacies of this process, especially for the generation of arylthio radical B.)
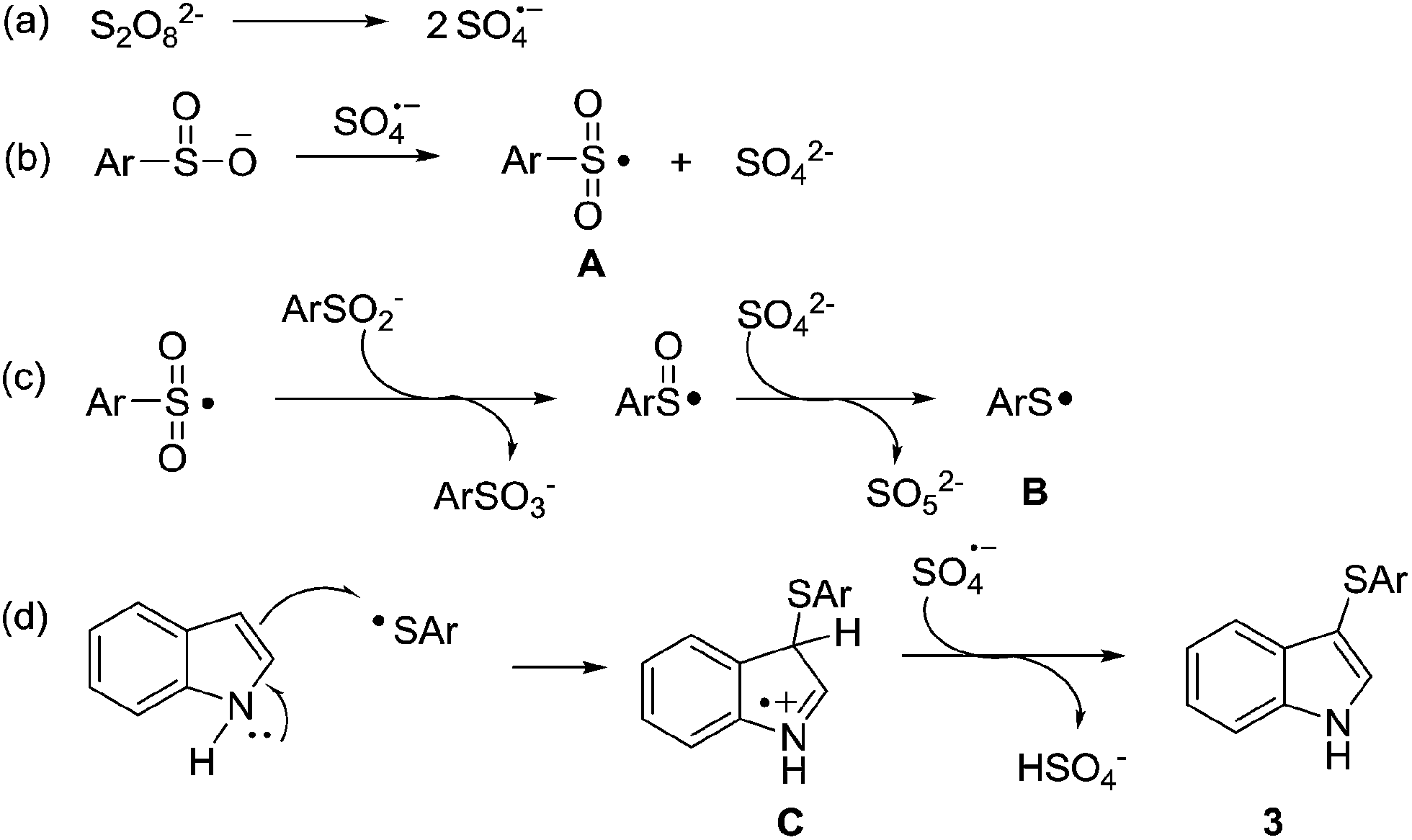 | ||
| Scheme 4 Proposed mechanism. | ||
In conclusion, we have developed a cheap, simple and efficient strategy for direct C3-sulfenylation of indoles. This strategy employs K2S2O8/arenesulfinate as the unprecedented thiolating system and CH3CN/H2O as the solvent without any other additives. It shows good tolerance towards carbon–halogen and aryl functionalities, thus promises further modifications of the desired 3-arylthioindoles. As such, the simplicity, high efficiency and environment-friendliness associated with this protocol suggest its potential for widespread use in the construction of pharmaceutically important molecules 3-arylthioindols. Further investigations to elucidate the detailed mechanism and synthetic applications of this efficient and practical sulfenylation protocol are currently underway in our lab.
Acknowledgements
We are grateful to the Beijing Natural Science Foundation (Grant no. 2144045), Beijing Municipal Education Commission Foundation (Grant no. KM201410028007), National Natural Science Foundation of China (Grant no. 21402128), Scientific Research Base Development Program of the Beijing Municipal Commission of Education, and Capital Normal University for support of our research.Notes and references
- (a) P. N. Craig, in Comprehensive Medicinal Chemistry, ed. C. J. Drayton, Pergamon, New York, 1991, vol. 8 Search PubMed; (b) M. Negwer, Organic Drugs and Their Synonyms: An International Survey, Akademic Verlag, Berlin, 7th edn, 1994 Search PubMed; (c) E. C. Taylor, in The Chemistry of Heterocyclic Compounds, ed. J. E. Saxton, Wiley-Interscience, New York, 1994 Search PubMed; (d) R. J. Sundberg, Indoles, Academic Press, New York, 1996 Search PubMed; (e) T. Kawasaki and K. Higuchi, Nat. Prod. Rep., 2005, 22, 761 RSC; (f) N. Saracoglu, Top. Heterocycl. Chem., 2007, 11, 145–178 Search PubMed.
- (a) J. J. Li and G. W. Gribble, in Palladium in Heterocyclic Chemistry, ed. J. E. Baldwin and F. R. S. M. R. Williams, Pergamon Press, New York, 2000, vol. 20, pp. 73–181 Search PubMed; (b) A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, Oxford, 3rd edn, 2010 Search PubMed; (c) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed; (d) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (e) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449 RSC , for selected examples on the construction and chemical modification of indoles, see:; (f) Z. Shi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572 CrossRef CAS PubMed; (g) S. Kirchberg, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2009, 48, 4235 CrossRef CAS PubMed; (h) S. Beaumont, V. Pons, P. Retailleau, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2010, 49, 1634 CrossRef CAS PubMed; (i) W. Wu, J. Xu, S. Huang and W. Su, Chem. Commun., 2011, 47, 9660 RSC; (j) L. Jiao and T. Bach, J. Am. Chem. Soc., 2011, 133, 12990 CrossRef CAS PubMed; (k) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750 CrossRef CAS PubMed; (l) Y. Wei, I. Deb and N. Yoshikai, J. Am. Chem. Soc., 2012, 134, 9098 CrossRef CAS PubMed; (m) W.-L. Chen, Y.-R. Gao, S. Mao, Y.-L. Zhang, Y.-F. Wang and Y.-Q. Wang, Org. Lett., 2012, 14, 5920 CrossRef CAS PubMed; (n) V. Pirovano, D. Facoetti, M. Dell'Acqua, E. D. Fontana, G. Abbiati and E. Rossi, Org. Lett., 2013, 15, 3812 CrossRef CAS PubMed.
- (a) G. De Martino, G. La Regina, A. Coluccia, M. C. Edler, M. C. Barbera, A. Brancale, E. Wilcox, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2004, 47, 6120 CrossRef CAS PubMed; (b) G. La Regina, M. C. Edler, A. Brancale, S. Kandil, A. Coluccia, F. Piscitelli, E. Hamel, G. De Martino, R. Matesanz, J. F. Diaz, A. I. Scovassi, E. Prosperi, A. Lavecchia, E. Novellino, M. Artico and R. Silvestri, J. Med. Chem., 2007, 50, 2865 CrossRef CAS PubMed.
- (a) T. M. Williams, T. M. Ciccarone, W. S. Saari, J. S. Wai, W. J. Greenlee, S. K. Balani, M. E. Goldman, J. M. Hoffman Jr, W. C. Lumma Jr, J. R. Huff, C. S. Rooney, P. E. Sanderson and A. D. Theoharides, PCT Int. Appl. WO 9419321, 1994; (b) R. Ragno, A. Coluccia, G. La Regina, G. De Martino, F. Piscitelli, A. Lavecchia, E. Novellino, A. Bergamini, C. Ciaprini, A. Sinistro, G. Maga, E. Crespan, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 3172 CrossRef CAS PubMed.
- (a) P. C. Unangst, D. T. Connor, S. R. Stabler, R. J. Weikert, M. E. Carethers, J. A. Kennedy, D. O. Thueson, J. C. Chestnut, R. L. Adolphson and M. C. Conroy, J. Med. Chem., 1989, 32, 1360 CrossRef CAS; (b) R. E. Armer and G. M. Wynne, PCT Int. Appl. WO 2008012511, 2008.
- C. D. Funk, Nat. Rev. Drug Discovery, 2005, 4, 664 CrossRef CAS PubMed.
- F. J. Lopez-Tapia, L. E. Lowrie Jr and D. D. Nitzan, PCT Int. Appl. WO 2008055847, 2008.
- Y. Maeda, M. Koyabu, T. Nishimura and S. Uemura, J. Org. Chem., 2004, 69, 7688 CrossRef CAS PubMed.
- M. Tudge, M. Tamiya, C. Savarin and G. R. Humphrey, Org. Lett., 2006, 8, 565 CrossRef CAS PubMed.
- (a) J. S. Yadav, B. V. S. Reddy, Y. J. Reddy and K. Praneeth, Synthesis, 2009, 1520 CrossRef CAS PubMed; (b) X. L. Fang, R. Y. Tang, P. Zhong and J. H. Li, Synthesis, 2009, 4183 CAS.
- C. C. Silveira, S. R. Mendes, L. Wolf and G. M. Martins, Tetrahedron Lett., 2010, 51, 2014 CrossRef CAS PubMed.
- (a) Z. Li, J. Hong and X. Zhou, Tetrahedron, 2011, 67, 3690 CrossRef CAS PubMed; (b) Z. Li, L. Hong, R. Liu, J. Shen and X. Zhou, Tetrahedron Lett., 2011, 52, 1343 CrossRef CAS PubMed.
- Y.-J. Guo, R.-Y. Tang, J.-H. Li, P. Zhong and X.-G. Zhang, Adv. Synth. Catal., 2009, 351, 2615 CrossRef CAS.
- M. Chen, Z. Huang and Q. Zheng, Chem. Commun., 2012, 48, 11686 RSC.
- For those with sulfenyl halides as electrophiles, see: (a) M. Raban and L. Chern, J. Org. Chem., 1980, 45, 1688 CrossRef CAS; (b) P. Hamel, J. Org. Chem., 2002, 67, 2854 CrossRef CAS PubMed , for quinone mono-O,S-acetals, see:; (c) M. Matsugi, K. Murata, H. Nambu and Y. Kita, Tetrahedron Lett., 2001, 42, 1077 CrossRef CAS; (d) M. Matsugi, K. Murata, K. Gotanda, H. Nambu, G. Anilkumar, K. Matsumoto and Y. Kita, J. Org. Chem., 2001, 66, 2434 CrossRef CAS PubMed , for N-thioimides, see:; (e) S. Ranjit, R. Lee, D. Heryadi, C. Shen, J. Wu, P. Zhang, K. Huang and X. Liu, J. Org. Chem., 2011, 76, 8999 CrossRef CAS PubMed; (f) E. Marcantoni, R. Cipolletti, L. Marsili, S. Menichetti, R. Properzi and C. Viglianishi, Eur. J. Org. Chem., 2013, 132 CrossRef CAS , for sulfonium salts, see:; (g) A. Deavin and C. Rees, J. Chem. Soc., 1961, 4970 RSC; (h) R. Ballini, E. Marcantoni and M. Petrini, Tetrahedron, 1989, 45, 6791 CrossRef CAS , for arylsulfonyl halides, see:; (i) Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188 RSC.
- D. Y. Huang, J. X. Chen, W. X. Dan, J. C. Ding, M. C. Liu and H. Y. Wu, Adv. Synth. Catal., 2012, 354, 2123 CrossRef CAS.
- (a) L. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307 RSC; (b) Z. Gao, X. Zhu and R. Zhang, RSC Adv., 2014, 4, 19891 RSC; (c) P. Sang, Z. Chen, J. Zou and Y. Zhang, Green Chem., 2013, 15, 2096 RSC.
- Ch. D. Prasad, S. Kumar, M. Sattar, A. Adhikary and S. Kumar, Org. Biomol. Chem., 2013, 11, 8036 CAS.
- (a) W. L. Ge and Y. Y. Wei, Green Chem., 2012, 14, 2066 RSC; (b) J. B. Azeredo, M. Godoi, G. M. Martins, C. C. Silveira and A. L. Braga, J. Org. Chem., 2014, 79, 4125 CrossRef CAS PubMed.
- F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS PubMed.
- F. Xiao, H. Xie, S. Liu and G.-J. Deng, Adv. Synth. Catal., 2014, 356, 364 CrossRef CAS.
- P. Katrun, S. Hongthong, S. Hlekhlai, M. Pohmakotr, V. Reutrakul, D. Soorukram, T. Jaipetch and C. Kuhakarn, RSC Adv., 2014, 4, 18933 RSC.
- Z. V. Todres, in Ion-Radical Organic Chemistry: Principles and Applications, CRC Press, New York, 2nd edn, 2009 Search PubMed.
- (a) H. H. Rao, X. Y. Ma, Q. Z. Liu, S. L. Cao and C.-J. Li, Adv. Synth. Catal., 2013, 355, 2191 CrossRef CAS; (b) X. Y. Ma, Z. F. Li, F. J. Liu, S. L. Cao and H. H. Rao, Adv. Synth. Catal., 2014, 356, 1741 CrossRef CAS.
- A reaction mixture of 2-methylindole (0.2 mmol), 3-methylindole (0.2 mmol), K2S2O8 (0.2 mmol, 1.0 equiv.) and p-tolylsulfinate (0.3 mmol, 1.5 equiv.) in CH3CN/H2O (1.0 mL, v/v ratio 10:1) was stirred at 100 °C under N2 atmosphere. In 24 h, product 2-methyl-3-p-tolylthioindole (3h) was isolated in 87%, while product 3-methyl-2-p-tolylthioindole (3q) in less than 5%.
- C.-M. Hu and F.-L. Qing, J. Org. Chem., 1991, 56, 6348 CrossRef CAS.
- F. Freeman, Chem. Rev., 1984, 84, 117 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedure for synthesis, characterization data, and 1H and 13C NMR spectra of compounds. See DOI: 10.1039/c4ra08669d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |