
Polylactide-b-poly(ethylene-co-butylene)-b-polylactide thermoplastic elastomers: role of polylactide crystallization and stereocomplexation on microphase separation, mechanical and shape memory properties†
Abstract
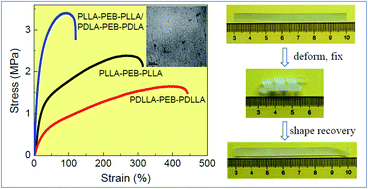
Polylactide-b-poly(ethylene-co-butylene)-b-polylactide (PLA–PEB–PLA) triblock copolymers containing PLA segments with different stereo-regularities such as poly(L-lactide) (PLLA), poly(D-lactide) (PDLA), and poly(D,L-lactide) (PDLLA) were prepared via the ring-opening polymerization of various lactides using α,ω-dihydroxy PEB as the macromolecular initiator. Molecular weight and chemical composition of copolymers were adjusted by changing the monomer-to-initiator ratio. Morphological, thermal, mechanical, and shape memory behaviors of PLA–PEB–PLA were explored. As confirmed by small angle X-ray scattering (SAXS) and transmission electrical microscopy (TEM), PLA–PEB–PLA adopted ordered microphase-separated morphology, depending on the copolymer composition and crystallizability of PLA segments. Spherical, hexagonally packed cylindrical, and lamellar structures were observed in PLA–PEB–PLA upon increasing the volume fraction of PLA. However, the morphological order was diminished in PLLA–PEB–PLLA/PDLA–PEB–PDLA enantiomeric blends, due to the preferential stereocomplexation of PLLA and PDLA segments before microphase separation. PLA–PEB–PLA showed the properties of thermoplastic elastomers. Their Young's modulus and tensile strength increased while the strain at break decreased upon increasing the fraction of PLA hard segments or with the crystallization or stereocomplexation of PLA domains. Interestingly, PLA–PEB–PLA elastomers showed shape memory behavior, which could be controlled by the crystallizability of PLA hard segments.
 Please wait while we load your content...
Please wait while we load your content...

