
A lithium/polysulfide semi-solid rechargeable flow battery with high output performance†
Kang Donga,
Shengping Wang*a and
Jingxian Yub
aFaculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, P. R. China. E-mail: spwang@cug.edu.cn
bSchool of Chemistry and Physics, The University of Adelaide, Adelaide, SA 5005, Australia
First published on 19th September 2014
Abstract
The feasibility of a semi-solid flow battery with polysulfide as catholyte is demonstrated, which gives a power density of 1.823 mW cm−2 at 4 mA cm−2. Compared to Li–S batteries with sulfur as cathode, the feasibility and flexibility using polysulfide as catholyte in flow-through mode create new potential for the practical application of conventional Li–S batteries.
The rapid exhaustion of fossil fuel resources and the need to limit greenhouse gas emissions urge researchers to develop new renewable energy resources to meet the urgent and increasing demands of portable electronics, electric vehicles (EV) and large-scale energy storage.1–4 Lithium–sulfur (Li–S) batteries, with a high theoretical energy density of 2600 W h kg−1, which is about 5 fold higher than the traditional Li-ion batteries based on intercalation electrodes, are regarded as one of the most promising candidates for the next generation of high-energy rechargeable batteries.5–7 In addition, the advantages of low-cost, environmental friendliness and wide operating temperature range are also appealing for large-scale energy storage.
However, Li–S batteries have been restricted from further commercialization by several hindrances,8,9 including (1) low utilization of active material due to the inherent poor electrical conductivity of sulfur and the fully discharged product Li2S; (2) low coulombic efficiency and rapid capacity fading mainly because of the high solubility of the intermediate polysulfides (i.e., Li2Sx, x = 8, 6, 4, and 3) in electrolyte and the shuttle of polysulfides (PS), which refers the long-chain polysulfides generated during the charging process can travel to the lithium anode and be reduced to short-chain polysulfides, which can then diffuse back to the cathode and be re-oxidized to long-chain polysulfides;10,11 (3) large volume expansion (∼80%) of sulfur and the deposition of Li2S2 and Li2S on the electrodes which may lead the cracking or disintegration of electrodes and the degradation of the overall cell performance.
To overcome these issues, numerous strategies have been developed to improve the discharge capacity, cyclability, and coulombic efficiency. Various approaches such as carbon matrix,5,9,12–16 surface coating,17,18 novel electrolytes19,20 and carbon interlayer21,22 have been used to confine sulfur in the cathode and thus improve the discharge capacity and cyclability. In addition, with the addition of LiNO3 in the electrolyte, a passivation layer can form on Li metal, which can prevent the “shuttle phenomenon” and improve the coulombic efficiency.23,24 Improvements have been achieved on the electrochemical performance of Li–S batteries, particularly on the decay of the discharge capacity during cycling. However, it must be admitted that most of the reported capacities have low sulfur loading of no more than 2 mg cm−2 in the cathode. It to some extent offsets its high energy density, which is regarded as the most prominent advantage of Li–S batteries.
Considering the fact that the diffusion of polysulfides from the confined matrixes is unavoidable and it is even essential for sulfur cathode. Therefore, polysulfide as cathode active material for Li/PS battery was proposed.25–27 Cui Yi et al. reported a Li/PS battery with appropriate amount of polysulfide infiltrating into carbon paper and gave a proof-of-concept trial by using a narrow voltage window to avoid the volume expansion for polysulfide's further large scale energy storage.25,28 Compared to the previous reported literatures, a semi-solid polysulfide flow cell (SSPFC) was introduced here, which also employs polysulfides but in flow-through mode, giving more realistic response. As shown in Fig. 1a, a laboratory-scale cell was designed and constructed employing carbon as the reaction matrix of cathode with polysulfides as catholyte continually flowing through its surface. Herein, we give a brief but systematic study on the electrochemical performances, reaction kinetics, and morphology of electrodes of a semi-solid polysulfide flow cell in flow-through mode. Polysulfide catholytes (see ESI† for experimental details) with different concentrations and nominal molecular formulas of Li2S6 and Li2S8 were also studied.
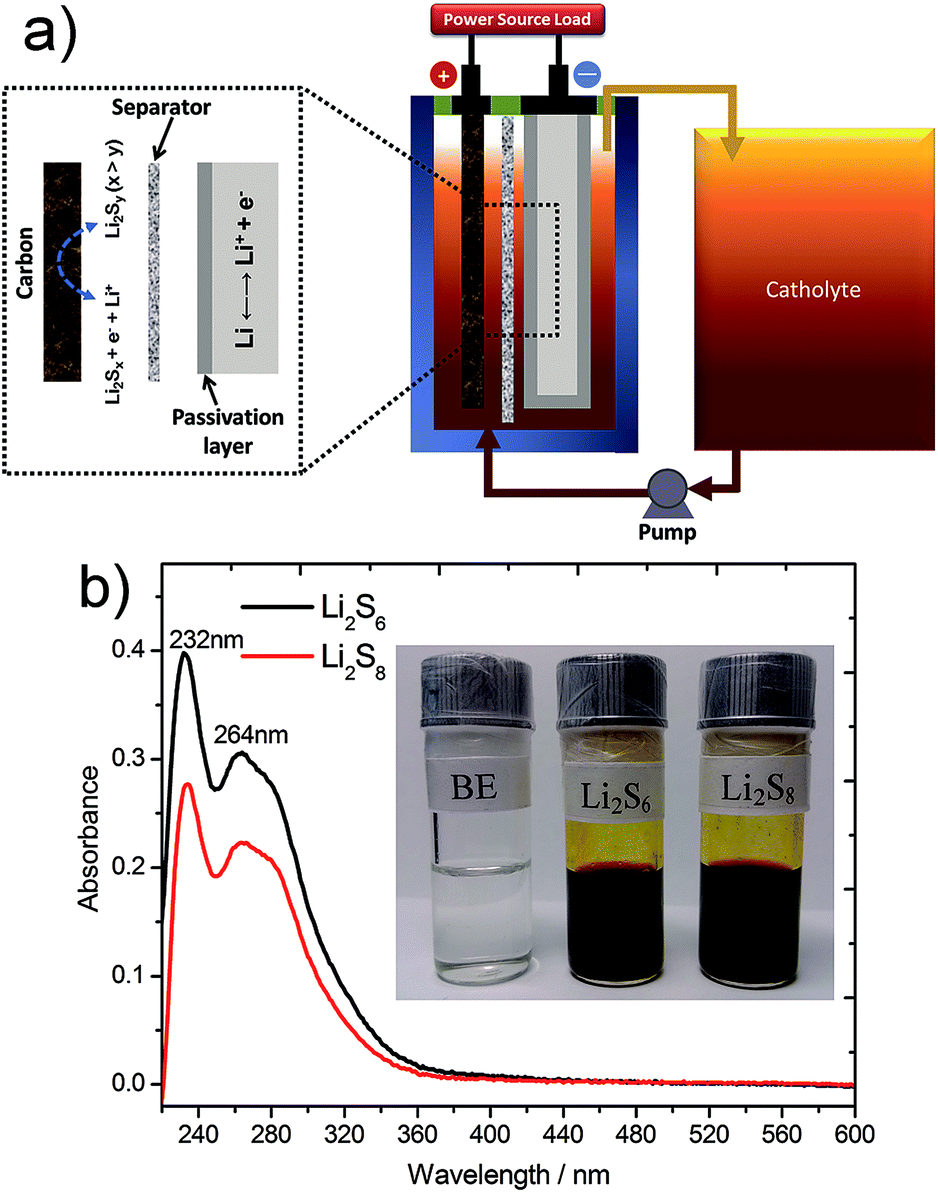 | ||
| Fig. 1 (a) Schematic diagram of the lithium/polysulfide semi-solid flow cell. (b) UV spectra of 0.2 M Li2S6 and 0.2 M Li2S8 catholytes using blank electrolyte as reference. Inset: the photographs of blank electrolyte, 0.2 M Li2S6 and 0.2 M Li2S8. | ||
A schematic of lithium/semi-solid polysulfide flow cell is shown in Fig. 1a. Lithium plates are employed as anode and lithium polysulfide solution is used as catholyte with carbon as the reaction matrix of cathode. A Li/Li symmetric cell with blank electrolyte (BE) stored at 25 °C was used to investigate the solid electrolyte interface (SEI) growth on the Li electrode (ESI, Fig. S1†). The passivation resistance of the Li electrode gradually increases with the storage time. In addition, the effect of the LiNO3 additive was examined by the energy-dispersive X-ray spectroscopy (ESI, Fig. S2†), which further demonstrated that an insoluble oxide passivation layer had generated on the surface of Li electrodes.23,29 It is worth noting that the passivation layer not only protects the lithium from the chemical reaction with polysulfides and the “shuttle phenomenon” but also makes it possible to build the SSPFC. This protection effectively improves the utilization of the active materials on both sides of the electrodes and the cycling efficiency, avoiding the tremendous waste of energy during the charging progress in practical application for the SSPFC.
The UV spectroscopy results are displayed in Fig. 2b. As the Gibbs free-energy of each polysulfide anion are so close that different types of polysulfides are co-existent in the solution through a series of reversible disproportionation reactions (eqn (1)–(3) in ESI†).30 A pure polysulfide solution with single polysulfide chain length cannot be obtained. Therefore, the solution in the form of Li2S8 and Li2S6 is likely to be a compound of several types of polysulfides and dissolved sulfur, corresponding to the peaks (Fig. 1b) around 232 and 264 nm.31,32
 | ||
| Fig. 2 (a) Cell potential versus current density at different current densities. (b) The galvanic power density and electrolytic power density versus current density. | ||
The proper electrochemical window was prior investigated by cyclic voltammetry using BE cycling on carbon cathode to avoid the undesired reduction of the electrolyte (Fig. S4†). The working potential window should be limited between 1.8 V to 3.2 V to avoid the reduction of LiNO3 around 1.5 V.33 The potential–current response (Fig. 2a) and the power–current relationship (Fig. 2b) of four types of polysulfide catholytes at different current densities were studied.
As shown in Fig. 2a, the open-circuit potential (OCP) of all the four lithium polysulfide catholytes are about 2.37 V, corresponding to the first discharge plateau and the generation of the long chain polysulfides in Li–S batteries. The potential deviate from the equilibrium potential upon current is applied to the battery, which is mostly due to the electrochemical polarization. With the increase of the current density, the potential of the battery give good linear response. Compared to the linear increase at the charge progress, the slight shift of the discharge potential of 0.05 M Li2S8 catholyte at high current density must be ascribed to the concentration polarization due to its relatively low concentration. The corresponding output and electrolytic power are shown in Fig. 2b. In the galvanic direction, the 0.2 M Li2S8 catholyte can offer the power density of 1.823 mW cm−2 (for the 30 μm thick carbon electrode) at 4 mA cm−2 and obviously, the output power densities can be further improved by increasing the thickness of the carbon electrode or using carbon with higher specific surface area.
As mentioned previously, there are a series of disproportionation reactions between S8, Li2S6, Li2S8 and Li2S. In addition, the insoluble S8 and Li2S are unstable in the polysulfides catholyte, which could react with soluble polysulfide as shown in eqn (4) and (5) in ESI.† Hence, with a lot of long chain polysulfides existing in the catholyte and continuous updating on the cathode's surface, the reaction at the cathode electrode could still be maintained within the liquid phase region, even though some solid S8 and Li2S may generate at deep depth of charge and discharge at high current density. The initial cyclic voltammograms of the Li/dissolved polysulfide cell at a narrow window between 2.5 and 2.2 V are shown in Fig. 3a. The two types of Li2S6 and Li2S8 prepared by different stoichiometric amounts both give an oxidation peak at 2.26 V and a deoxidization peak around 2.36 V. This couple of reversible redox peaks corresponds to the reaction between S8 and the linear high order lithium polysulfides (e.g. Li2S6 and Li2S8).
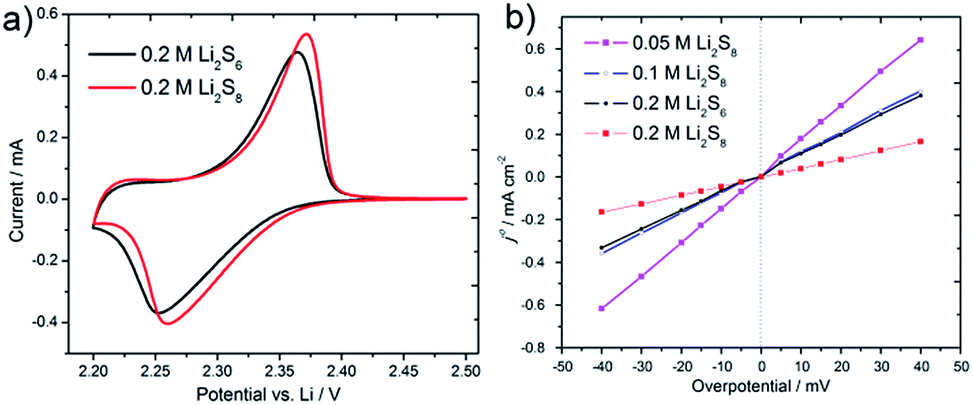 | ||
| Fig. 3 (a) Cycle voltammograms of Li/dissolved polysulfide cell at a sweep rate of 0.1 mV s−1 using a type of Swagelok-type cell. (b) The polarization curves of four kinds of lithium polysulfide catholytes. | ||
Low overpotential was further applied to the SSPFC to compare the reaction kinetics of the four PS catholytes. The polarization curves (Fig. 3b) indicate a good linear relation around the equilibrium potential due to the low overpotential and the continuous update of PS on the surface of cathode. During the weak polarization region, the polarization resistance can be easily obtained through the slopes of the polarization curves. It is obviously to find that 0.2 M Li2S8 had the minimum polarization resistance (Rp = 243.1 Ohm cm−2) and thus had the maximum exchange current density j0, while 0.05 M Li2S8 had the minimum j0. The large j0 of 0.2 M Li2S8 reflects a fast kinetic during the charge and discharge progress, which indicates a good electrochemical performance at high current density and a bright future for its application in the lithium/polysulfide semi-solid flow cell.
Fig. 4a displays the charge and discharge profiles of Li2S6 polysulfide at different current densities. All the voltage profiles remained steady and do not show any obviously decline even after several one-hour discharges at high current density, which implies a steady output power under a certain current density. At low current densities, the voltage profiles are almost straight lines parallel to the X axis. But when the current density is higher than 1 mA cm−2, there is a slight voltage drop at the initial discharge stages and the voltage fluctuation appear during the charge stages as shown in Fig. 4a, which should be attributed to the large concentration polarization due to the severe consumption of Li2S6 and the rapid accumulation of product. The voltage fluctuation can be further optimized by optimizing the flow rate, the concentration of the PS catholyte as well as the stirring velocity in the cell. After the voltage undergoes a short-time decline at the initial stage of discharge progress, the discharge voltage keep steady for the subsequent about 55 minutes, which indicates a superior electrochemical performance of the lithium/polysulfide semi-solid flow cell.
 | ||
| Fig. 4 (a) The voltage profile of 0.2 M Li2S6 catholyte cycling for long time at different current densities; (b) the voltage profiles and energy efficiency of 0.2 M Li2S6 catholyte cycling at 2.5 mA cm−2 for 50 times; to show clearly, only 20 cycles of the voltage profiles are given. (c) The energy efficiency and comparison of the initial 3 cycles (open circles) and final 3 cycles (line) of 0.2 M Li2S8 catholyte cycling at 2.5 mA cm−2 for 200 times. (d) SEM images of carbon cathode before and after cycling. The scale bar in (d) equals 500 nm. | ||
Further efforts were put on the cycling performance of the constant-current. Curves of 0.2 M Li2S6 and 0.2 M Li2S8 catholyte cycling at short time (10 minutes for charge and discharge, respectively) but high current density (2.5 mA cm−2) are shown in Fig. 4b and 4c. With the limit of the charge and discharge time, the voltage profiles and the cutoff of the voltage are almost same during 50 cycles. Cycling performance for 200 cycles and some selected voltage profiles are shown in Fig. 4c. Compared to the initial 3 cycles, the voltage profiles of the final 3 cycles do not show obviously changes except for the decrease of the voltage platform, which should be mainly attributed to the constant growth of the passivation layer on the Li electrode. Although the presence of the passivation layer could protect lithium from the possible chemical reaction and suppress the shuttle phenomenon and thus makes it possible to build the SSPFC, the composition and thickness of the passivation layer should be further optimized e.g. by controlling the concentration of LiNO3 in catholyte. Extended cycling performance for 500 cycles is displayed in Fig. S5.† Moreover, the steady energy efficiency of the 0.2 M Li2S6 and 0.2 M Li2S8 demonstrate the good reversibility of the SSPFC, which should be partly ascribed to the positive effect of LiNO3 on suppressing the shuttle phenomenon. In addition, unlike Li-insertion compounds and the conventional lithium sulfur batteries, the lithium/polysulfide semi-solid flow cell would not cause a severe volume change of the cathode by controlling the depth of reaction. Obvious morphology changes of the carbon matrix before and after cycling, as shown in Fig. 4d, could not be observed, which further indicates a good stability of the cathode structure during the cycling.
Therefore, with polysulfide in flow-through mode, the predominant electrochemical reactions could be confined in liquid phase region, which can nearly solve the challenges that facing Li–S batteries, including the poor electrical conductivity, large volume expansion and rapid capacity fading mostly due to the dissolution of polysulfides. The SSPFC combines the high energy density of Li–S batteries with the modularity and flexible operation of flow cell. Moreover, compared to conventional lithium sulfur batteries using sulfur as the cathode active material, energy is stored in the flowing polysulfide catholyte and the output energy is no longer limited by the sulfur loading on the cathode.
Conclusions
In summary, the feasibility of lithium/polysulfide semi-solid flow cell utilizing soluble polysulfides solution as catholytes in flow-through mode have been demonstrated, which could offer a power density of 1.823 mW cm−2 at 4 mA cm−2. The solubility, cycling stability and reversibility of polysulfides bring hope to the development of lithium/polysulfide semi-solid flow cell. However, it should be emphasized that the cell can be further improved. Optimization of the material of cathode, the concentration and composition of the catholyte, as well as the design and operating parameters could lead to significant performance improvements in the future. The semi-solid polysulfide flow cell proposed here could create new potential for the commercialization of conventional Li–S batteries and provide a wide range of opportunities to be applicable for cost-effective, large-scale energy storage.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21173198) and the Natural Science Foundation of Hubei Province, China (2013BHE014). The authors thank the Materials and Chemistry Analytical and Testing Center of CUG for SEM, EDS and UV measurements.Notes and references
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed
- R. Van Noorden, Nature, 2014, 507, 26–28 CrossRef CAS PubMed
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed
- Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed
- X. Ji and L. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS PubMed
- X. G. Sun, X. Wang, R. T. Mayes and S. Dai, ChemSusChem, 2012, 5, 2079–2085 CrossRef CAS PubMed
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed
- N. Li, M. Zheng, H. Lu, Z. Hu, C. Shen, X. Chang, G. Ji, J. Cao and Y. Shi, Chem. Commun., 2012, 48, 4106–4108 RSC
- J. Jin, Z. Wen, G. Ma, Y. Lu, Y. Cui, M. Wu, X. Liang and X. Wu, RSC Adv., 2013, 3, 2558–2560 RSC
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed
- B. Ding, C. Yuan, L. Shen, G. Xu, P. Nie and X. Zhang, Chem.–Eur. J., 2013, 19, 1013–1019 CrossRef CAS PubMed
- F. Wu, J. Z. Chen, R. J. Chen, S. X. Wu, L. Li, S. Chen and T. Zhao, J. Phys. Chem. C, 2011, 115, 6057–6063 CAS
- Z. W. Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331–1336 CrossRef PubMed
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481–1489 CrossRef PubMed
- R. Xu, I. Belharouak, J. Li, X. Zhang, I. Bloom and J. Bareño, Adv. Energy Mater., 2013, 3, 833–838 CrossRef CAS
- Y.-S. Su, Y. Fu, T. Cochell and A. Manthiram, Nat. Commun., 2013, 4, 2985–2992 Search PubMed
- G. Zhou, S. Pei, L. Li, D.-W. Wang, S. Wang, K. Huang, L.-C. Yin, F. Li and H.-M. Cheng, Adv. Mater., 2013, 26, 625–631 CrossRef PubMed
- D. Aurbach, E. Pollak, R. Elazari, G. Salitra, C. S. Kelley and J. Affinito, J. Electrochem. Soc., 2009, 156, A694–A702 CrossRef CAS PubMed
- Y. V. Mikhaylik, I. Kovalev, R. Schock, K. Kumaresan, J. Xu and J. Affinito, ECS Trans., 2010, 25, 23–34 CAS
- Y. Yang, G. Zheng and Y. Cui, Energy Environ. Sci., 2013, 4, 1552–1558 Search PubMed
- Y. Fu, Y.-S. Su and A. Manthiram, Angew. Chem., 2013, 125, 7068–7073 CrossRef
- S. S. Zhang and J. A. Read, J. Power Sources, 2012, 200, 77–82 CrossRef CAS PubMed
- M. Agostini, D.-J. Lee, B. Scrosati, Y. K. Sun and J. Hassoun, J. Power Sources, 2014, 265, 14–19 CrossRef CAS PubMed
- Y. V. Mikhaylik, US Pat., 2008/0193835 A1, 2008
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CrossRef CAS PubMed
- C. Barchasz, F. Molton, C. Duboc, J. C. Leprêtre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3973–3980 CrossRef CAS PubMed
- Y. Li, H. Zhan, S. Liu, K. Huang and Y. Zhou, J. Power Sources, 2010, 195, 2945–2949 CrossRef CAS PubMed
- S. S. Zhang, Electrochim. Acta, 2012, 70, 344–348 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra08413f |
| This journal is © The Royal Society of Chemistry 2014 |