
Preparation and characterization of TiO2–Graphene@Fe3O4 magnetic composite and its application in the removal of trace amounts of microcystin-LR†
Abstract
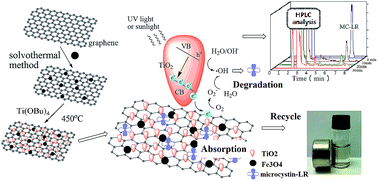
Microcystins (MCs), a family of potent cyclic heptapeptides, are produced by cyanobacteria blooms in eutrophic water and can cause acute and chronic toxicity and even mortality to animals and humans. Previous MC removal strategies concerned only highly contaminated water, in which the concentration of the pollutant was considerably larger than that in the natural world. Herein, we developed a ternary composite of TiO2-coated magnetic graphene and used it as an adsorbent and photocatalyst to efficiently remove microcystin-LR (MC-LR) from water. The two-dimensional sheets of graphene were decorated with a large quantity of spherical Fe3O4 nanoparticles (10–20 nm) and then coated with crystallized TiO2. These TiO2–graphene@Fe3O4 composites exhibited a high magnetic response to the external magnetic field. And the huge surface of the graphene dramatically boosted the adsorbability and charge mobility, which lowered the recombination rate of electron–hole pairs, and hence systematically enhanced photocatalytic activity. The combination of adsorption and photodegradation endowed the composite with a better performance in the removal of trace amounts of MC-LR than the commercial photocatalyst, Degussa P25. The concentration of MC-LR can be lowered to less than 1 μg L−1 (a provisional safety guideline by the World Health Organization) from 500 μg L−1 under UV light in 30 min. The loading of TiO2–graphene@Fe3O4, the pH, and the UV energy were also optimized. Moreover, the stable removal capability of TiO2–graphene@Fe3O4 was confirmed over multiple cycles. Finally, the removal performance was also evaluated under natural light illumination in real surface water samples. This work paves the way for the development of more efficient and easily separable purifiers for the removal of pollutants and toxins from contaminated water.
 Please wait while we load your content...
Please wait while we load your content...

