
A multi-photoresponsive molecular-hybrid for dual-modal photoinactivation of cancer cells
Aurore Fraix†
a,
Stefano Guglielmo†b,
Venera Cardilec,
Adriana C. E. Grazianoc,
Ruxandra Grefd,
Barbara Rolandob,
Roberta Fruttero*b,
Alberto Gascob and
Salvatore Sortino*a
aLaboratory of Photochemistry, Department of Drug Sciences, University of Catania, 95125 Catania, Italy. E-mail: ssortino@unict.it
bDepartment of Drug Science and Technology, University of Torino, 10125 Torino, Italy. E-mail: roberta.fruttero@unito.it
cDepartment of Bio-Medical Sciences, Physiology Division, University of Catania, 95125 Catania, Italy
dUMR CNRS 8612, Faculty of Pharmacy, Paris Sud University, 92290 Châtenay-Malabry, France
First published on 8th September 2014
Abstract
We report the design, synthesis, photochemical characterization and biological evaluation of a novel molecular conjugate in which two chromogenic centers, a porphyrin unit and a nitroaniline derivative, are covalently linked through an alkyl spacer. This molecular hybrid can be encapsulated in biocompatible, water soluble polymer nanoparticles where it shows satisfactory fluorescence emission and capability to generate simultaneously the cytotoxic singlet oxygen and nitric oxide upon excitation with visible light. The photoactive nanoassembly can be delivered to A375 melanoma cancer cells where it can be detected through its red fluorescence, and is capable of inducing amplified cell mortality by bimodal action due to the concomitant photoproduction of reactive oxygen and nitrogen oxygen species.
Introduction
Multimodal cancer therapies exploit either additive or synergistic effects arising from the generation of different active species for improving the therapeutic efficacy.1 In this frame, the spatio-temporal control of the bio-active agents represents a challenging objective since it allows, in principle, maximization of the therapeutic action and minimization of side effects.2 Light represents a powerful tool for the rapid introduction of active species in a biological environment, mimicking an “optical microsyringe” with exquisite control of three main factors such as site, timing and dosage, which are determining for the therapeutic outcome.3–5 In addition, photochemical triggering offers the great benefit of not perturbing the physiological values of parameters such as temperature, pH and ionic strength, fundamental prerequisite for biomedical applications. These unique features make the photoactivated systems a powerful arsenal in the burgeoning field of nanomedicine with intriguing potential to tackle cancer diseases in a noninvasive way.3–7Multidrug resistance (MDR), the major factor in the failure of many forms of chemotherapy,8 calls for a shift of attention to alternative treatment modalities. At this regard, the light-controlled generation of reactive oxygen species (ROS) and reactive nitrogen oxygen species (RNOS) such as singlet oxygen (1O2) and nitric oxide (NO) by using appropriate photochemical precursors represents a fascinating and unconventional strategy.
1O2 is the key species in photodynamic therapy (PDT), a well-established therapeutic modality for the treatment of malignant lesions in humans. It implies the combined use of visible light, a non-toxic photosensitizing dye (PS) and the molecular oxygen.9 The PS generally belongs to the class of porphyrins and phthalocyanines10 and is administered by intravenous, intraperitoneal or topical route. It accumulates preferentially into the tumor cells as compared with the normal cells and its intracellular distribution is depending on its structure.11 Illumination of the tumoral area with an opportune dose of appropriate wavelength light, leads to the population of the lowest and long-lived excited triplet state of the PS. The return of the system to the ground state occurs mainly by energy transfer to molecular oxygen, generating the highly reactive 1O2 which induces selective tumor cell and tumor tissue destruction through a number of mechanisms.12
Nitric oxide (NO) is an important gaseous endogenous messenger which plays a variety of roles in the human physiology and pathophysiology.13 NO contributes to maintain micro and macrovascular homeostasis inducing vasodilation, inhibition of platelet aggregation, modulation of platelet and leukocyte adherence to vessels, and inhibition of smooth muscle cell proliferation.14 In the central nervous system it is implicated in neural signaling, neurotoxicity, neuroprotection, synaptic plasticity and modulation of behavioral pathways.15 Peripherally NO modulates a number of reflexes by acting as neurotransmitter at the endings of non-adrenergic non-cholinergic nerves.15 NO is also one of the final effectors in the immune system, where it triggers tumoricidal, antimicrobial or antiparasitic actions.16 Indeed, NO plays important roles in the biology of tumors and in the inflammation processes.17,18 Therefore, great attention has been paid to products able to release NO under physiological conditions as potential drugs to fight a variety of diseases.19,20 NO-photodonors, namely molecules capable of producing NO under the action of the light, represent a particular class of these compounds.21,22 They allow an accurate control of the timing, location and dosage of NO-release. This aspect is very important since NO can be therapeutic or toxic (double edged sword) depending on the doses.23
The combination of 1O2 and NO represents a very appealing strategy in view of multimodal therapeutic systems. 1O2 and NO share in fact several important features such as: (i) small size and absence of charge, (ii) capability to attack biological substrates of different nature (i.e., lipids, proteins, and DNA), (iii) absence of multidrug resistance, (iv) confinement of their action to short distances from the production site inside the cells (<20 nm for 1O2 and <200 μm for NO), due to their short lifetime, reducing systemic toxicity issues common to many conventional drugs. Besides, since NO photorelease is independent from O2 availability, it very well complements PDT at the onset of hypoxic conditions, typical for some tumors, where PDT may fail.
The visualization of the phototherapeutic precursor in a cellular environment through fluorescence techniques represents an indispensable requisite in view of image-guided cancer phototherapy. This has recently led to the general term “photosensitizer fluorescence detection” in reference to all applications in which a PS is also used to generate fluorescence contrast.24 Therefore, the creation of single platforms enabling simultaneous photogeneration of 1O2 and NO and, at the same time, tracing in a cellular environment via fluorescence techniques is very challenging. We have recently reported a number of multifunctional nanoconstructs with imaging and bimodal therapeutic modalities by the non-covalent assembling of suitable PSs for PDT and tailored NO photodonors.25 On these grounds, the achievement of molecular hybrids in which a PS for PDT and a NO photodonor are covalently connected without affecting the photochemical behaviour of the individual components, would represent a significant step forward with respect to the above non-covalent systems. These last can potentially suffer disassembling after penetration in the cellular environment and, as a consequence, photogenerate the cytotoxic agents in different sites of the cell compartment. On the other hand, molecular conjugates, would offer in principle the great advantage of a much more precise control of timing, location and dosage of the cytotoxic species. In fact, the covalent connection of the photoactive precursors ensures that the photodelivery events occur exactly in the “very same region of space” of the cell component. To this end, we have developed herein the novel multiphotoresponsive molecular hybrid 1 (Scheme 1) in which a porphyrin unit and a tailored NO photodonor are integrated within the same molecular skeleton through an alkyl spacer. In this contribution we report the synthesis, photochemical characterization and biological evaluation of this molecular conjugate and the related model compounds 2 and 3. In particular, we show that the conjugate 1 (i) can be encapsulated within nanoparticles of the biocompatible and water soluble copolymer 4 used as suitable carrier system, (ii) photogenerates simultaneously 1O2 and NO under visible light stimuli, (iii) exhibits satisfactory red fluorescence allowing the visualization of its localization in melanoma cancer cell, (iv) induces amplified cell mortality by bimodal photoaction (Scheme 1).
 | ||
| Scheme 1 Molecular structures of the conjugate 1, the model compounds 2 and 3, and the polymer carrier 4. | ||
Results and discussion
Design and synthesis
In search of a viable strategy to construct a molecular hybrid able to display fluorescence properties associated to the simultaneous photogeneration of 1O2 and NO we have deliberately chosen a nitroaniline derivative bearing a CF3 substituent in the ortho position with respect to the nitro group and a tetra-pyridyl porphyrin as suitable chromogenic units, respectively. We have demonstrated that such a class of nitroaniline-like chromophores are suitable NO photodonors as they satisfy several prerequisites for bio-applications including excitation with visible light and formation of non-toxic side photoproducts.26,27 The twisted conformation adopted by the nitro group with respect to the aromatic plane is crucial for the NO photorelease. Similarly to other nitroaromatics having the same molecular conformation, the mechanism of NO release involves an initial nitro-to-nitrite photorearrangement followed by cleavage of the O–NO bond.28 Besides, pyridyl porphyrins derivatives are good red photoemitters, excellent 1O2 PS and, in addition, they can be ad-hoc derivatized via simple synthetic procedures.29The rationale behind the choice of the above chromogenic components has its root in our previous works in which we have demonstrated that the non-covalent assembling of porphyrin centers and nitroaniline-based NO photodonors in a restricted region of space after incorporation in micelles,30 nanoparticles31 and host–guest complexes,32 does not influence the photochemical behavior of both chromophores. Note that, in contrast to non-photoresponsive compounds, the preservation of the photobehavior of independent photoactive components after their covalent linking is not a “trivial result”. In most cases, the response to light of the single photoactive units located in a so close proximity can be considerably influenced, in both nature and efficiency, by the occurrence of competitive photoprocesses (i.e., photoinduced energy and/or electron transfer, non-radiative deactivation, etc.), which do not allow the individual components to work in parallel after light absorption. Taking into account these considerations, we have synthesized the conjugate 1 and the model compounds 2 and 3 according to the synthetic steps illustrated in Scheme 2. Reaction of 3c and of 1-bromopropane with an excess of porphyrin 1a in glacial acetic acid solution gave rise to the compounds 1 and 2 in poor yields, after flash chromatography purification. The products 3b and 3 were easily obtained by treatment of the commercial 4-chloro-1-nitro-2-(trifluoromethyl)benzene (3a) with methylamine and N-methylpropylamine respectively, in absolute ethanol solution in the presence of Na2CO3. Intermediate 3c was prepared by refluxing in acetonitrile 1,3-dibromopropane and 3b in the presence of K2CO3.
 | ||
| Scheme 2 Synthesis of the conjugate 1 and the model compounds 2 and 3. Reagents and conditions: (i) 3c, refluxing glacial CH3COOH, 20 h; (ii) 1-bromopropane, glacial CH3COOH, 120 °C (closed vessel), 20 h; (iii) CH3NH2 33% in absolute EtOH, Na2CO3, 70 °C, 36 h (closed vessel); (iv) 1,3-dibromopropane, K2CO3, refluxing CH3CN, 5 days; (v) N-methylpropylamine, Na2CO3, absolute EtOH, 70 °C, 36 h (closed vessel). | ||
Spectroscopic and photochemical properties
Fig. 1A shows the absorption and fluorescence emission spectra of the molecular hybrid 1 and the related model compounds in methanol solution. The absorption profile of 1 reflects fairly well that of an equimolar mixture of the model compounds 2 and 3, with an experimental error of about 10%, accounting for only a weak interaction between the two chromophoric units in the ground state. Besides, 1 exhibits the typical dual band fluorescence emission of the porphyrin moiety, analogously to that observed for an optically matched solution of the model compound 2. We obtained a fluorescence quantum yield Φf = 0.06 for 1 and 2, a value in the range reported for other pyridyl porphyrin derivatives.33 This result suggests that the porphyrin core retains its emissive properties after the covalent linkage with the nitroaniline appendage. | ||
| Fig. 1 (A) Absorption spectra of methanol solutions of 1 (a), 2 (b) and 3 (c). Fluorescence emission spectra (λexc = 505 nm) of methanol solutions of 1 (d) and 2 (e). [1] = [2] = [3] = 3.3 μM; T = 25 °C. (B) Absorption and fluorescence emission spectra of 1 in the presence of dispersions of 4 in aqueous phosphate buffer 10 mM pH 7.4 (a and c) and DMEM medium (b and d). The inset shows the hydrodynamic diameter for dispersions of 4 in the presence of 1 in aqueous phosphate buffer 10 mM pH 7.4 (a) and DMEM medium (b). [1] = 3.3 μM; [4] = 4 mg mL−1; 25 °C. | ||
Compound 1 is totally insoluble in both phosphate-buffered aqueous solution and in DMEM medium. However, compound 1 was slightly soluble in water containing 1% DMSO but unfortunately the presence of large aggregates precluded fluorescence. Focusing on solubility and aggregation issues in aqueous medium, cyclodextrin (CD) polymers offer the possibility of guest interaction with diverse binding sites, i.e. within the 3D macromolecular network and the CD cavities, thereby enhancing the apparent solubility and regulating the self-association tendency of drugs.34 On these basis we used polymer 4 (see Scheme 1) as a suitable nanocarrier. Polymer 4 consists of β-CD units interconnected by epichlorohydrin spacers to form glyceryl cross-linked β-CD polymer. This polymer is well tolerated in vivo35 and highly soluble in water where it exists under the form of nanoparticles (NPs) of ca. 25 nm in diameter.36 Due to the presence of different hydrophobic nanodomains these NPs are able to entrap a variety of guests34,37,38 with enhanced stability constants and payloads as compared with the unmodified β-CD.
Compound 1 becomes fairly soluble in the presence of aqueous dispersion of 4. As shown in Fig. 1B, both the absorption and the emission profiles (spectra a and c) were very similar to those observed in methanol confirming the effective encapsulation of 1 within 4, mainly under its monomeric form. The fluorescence quantum yield was even higher than in methanol solution, being Φf = 0.09. Dynamic light scattering measurements indicate the average hydrodynamic diameters of ca. 35 nm for the NPs entrapping 1 (a, inset Fig. 1B). This value is slightly larger than that observed for the empty NPs (ca. 25 nm)36 and in excellent agreement with that found for the same carrier entrapping chromophores of similar molecular sizes as 1.34,37,38 Interestingly, the absorption and emission characteristics (b and d in Fig. 1B) together with NPs sizes (b in the inset of Fig. 1B) were preserved quite well in DMEM medium where we obtained an emission quantum yield Φf = 0.07 and a hydrodynamic diameter of ca. 33 nm.
The excited triplet state of the porphyrins is the key transient intermediate for the photosensitization of 1O2 and its effective generation is thus crucial for the photodynamic action.9 Laser flash photolysis with nanosecond time-resolution is a powerful tool for obtaining spectroscopic and kinetic features of excited triplets of porphyrins since these transient species exhibit very intense absorptions in the visible region and possess lifetimes falling in the microsecond time scale.39 Fig. 2A shows that laser excitation of compound 1 in methanol results in the formation of a transient absorption with a maximum at ca. 450 nm and a bleaching due to the Soret ground-state absorption. This species which decays mono-exponentially with a lifetime of ca. 20 μs (inset Fig. 2A), is virtually the same to that obtained upon laser excitation of the model compound 2 (which decays with similar lifetime) and can be safely attributed to the lowest excited triplet state of the porphyrin core.39
 | ||
| Fig. 2 (A) Transient absorption spectra observed 0.1 μs after 532 nm laser excitation (E532 ∼ 12 mJ per pulse) of Ar-saturated and optically matched methanol solutions of 1 (■) and 2 (○). The inset shows the decay trace monitored at 450 nm and the related first-order fitting. (B) Transient absorption spectra observed 1 μs (■), 390 μs (○), 850 μs (●) and 1700 μs (□) after 532 nm laser excitation (E532 ∼ 12 mJ per pulse) of Ar-saturated aqueous dispersions of 4 in phosphate buffer 10 mM pH 7.4 in the presence of 1. The inset shows the decay trace monitored at 440 nm and the related first-order fitting. [1] = 3.3 μM; [4] = 4 mg mL−1; 25 °C. | ||
Analogously to what observed for the fluorescence emission, the presence of the nitroaniline-derivative appendage does not influence the efficiency of the triplet population. In fact, since the two samples were optically matched at the excitation wavelength and the laser energy used was the same, the intensity of the transient absorption is directly related to the triplet quantum yield ΦT. We obtained a value for ΦT of 0.85 (see Experimental section), in excellent agreement with other tetrapyridyl porphyrin derivatives.33
The triplet state is also effectively populated when 1 is encapsulated in the NPs of the polymer carrier 4 in aqueous solution. Fig. 2B shows the transient absorption spectra obtained under these conditions and recorded at different delay times with respect to the initial laser pulse. Apart from a slight blue shift in the absorption maximum, the spectrum taken at the shortest delay time shows a profile very similar to that observed in methanol solution. The time evolution of the absorption reveals that no new transient species is formed concurrently to the triplet decay, ruling out any possible reaction of this species with the NPs. The excited triplet state decays mono-exponentially with a triplet lifetime of ca. 1000 μs (inset Fig. 2B), more than one order of magnitude longer than that observed in the organic solvent. The lengthening of the triplet decay observed is quite common upon incorporation of chromophores hosted in systems such as micelles, liposomes and CDs and is usually related to the protection exerted by the host cage against external quenching impurities, to a perturbation of the inter-system crossing process to the ground state, or to both.40
Energy transfer from the triplet of 1 to molecular oxygen results in the photogeneration of 1O2. Time-resolved near-infrared luminescence with sub-microsecond time resolution is the most suitable technique to unequivocally demonstrate the generation of singlet oxygen, 1O2. This species, in fact, exhibits a typical luminescence signal at 1.27 μm with a lifetime ranging in the microseconds time-scale.41 We have examined the singlet oxygen produced by energy transfer from 1 and the model compound 2 to molecular O2, in partially deuterated (80% D2O) aqueous dispersions of the carrier 4. Fig. 3 shows the typical phosphorescence signal at 1270 nm observed for 1 and the model compound 2. In both cases, the kinetic analysis of the decay traces gives a lifetime of ca. 40 μs, consistent with the reported value in the solvent used.41
 | ||
| Fig. 3 Representative decay kinetic and related first-order fitting of 1O2 observed upon 532 nm laser excitation (E532 ∼ 12 mJ per pulse) of aqueous dispersion (80% D2O) of 4 in the presence of either 1 (A) or the model compound 2 (B). [1] = [2] = 3.3 μM; [4] = 4 mg mL−1; Phosphate buffer 10 mM, pH 7.4, 25 °C. | ||
Analogously to what observed for the precursor triplet state, the quantum yield of 1O2 (ΦΔ) for 1 was basically the same to that determined for the model 2. We obtained a value for ΦΔ = 0.55 (see Experimental) for both compounds, in excellent agreement to that reported for monocationic pyridyl porphyrins.29
The NO photorelease properties of the conjugate 1 are demonstrated by the direct and real-time monitoring of this transient species using an ultrasensitive NO electrode which directly detects NO with nM concentration sensitivity by an amperometric technique.42 The results illustrated in Fig. 4 provide evidence that the molecular hybrid 1 and the model compound 3 are stable in the dark but supply NO exclusively upon illumination with visible light. Note that, the rate for the NO photorelease was ca. 50 nM s−1 for both compounds suggesting that the porphyrin chromophore does not influence the capability of the NO photorelease in the conjugate.
 | ||
| Fig. 4 NO release profiles observed upon irradiation (λexc = 405 nm) of aqueous dispersion of 4 in the presence of either 1 (a) or the model compound 3 (b). [1] = [3] = 3.3 μM; [4] = 4 mg mL−1; phosphate buffer 10 mM, pH 7.4, 25 °C. The data are corrected for the different fraction of absorbed photons by 1 and the model compound 3, at the excitation wavelength. | ||
Cell imaging and viability assay
The suitability of the multifunctional molecular conjugate 1 for cell fluorescence imaging and bimodal phototoxicity was demonstrated by in vitro experiments performed with A375 cells, a human amelanotic melanoma cell line. The cell uptake of 1 was determined after 4 h of incubation of a DMEM suspension of the carrier 4 in the presence of 1, using fluorescence microscopy (Fig. 5). The typical porphyrin-associated fluorescence reveals that 1 is mainly accumulated in the cytoplasm of the melanoma cells, observed as red fluorescent spots, but no relevant nucleus associated fluorescence was observed. Fluorescence examination demonstrated the absence of obvious cell toxicity and of nuclear fragmentation, a marker of cell apoptosis, by DAPI staining.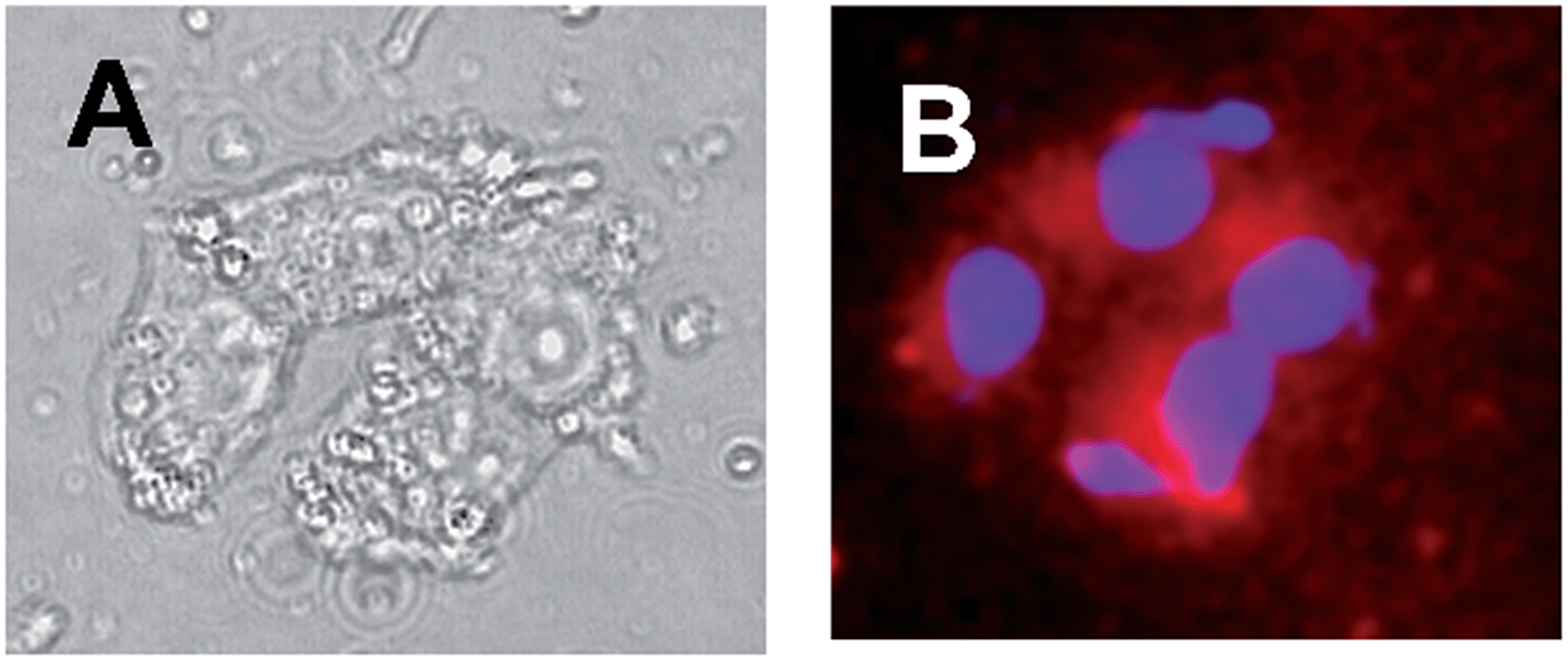 | ||
| Fig. 5 Evaluation of the uptake of 1 by A375 melanoma cells. (A) Transmission microscopy. (B) Merging images of the cells analysed with a porphyrin and a DAPI emission filters, respectively, indicating accumulation of 1 in cells (red spots). [1] = 3.3 μM; [4] = 4 mg mL−1; 25 °C. | ||
To validate the feasibility of using this new molecular hybrid for bimodal phototherapeutic activity, the melanoma cells were incubated under different experimental conditions and were either kept in the dark or irradiated with visible light in the range 400–800 nm. Cell cytotoxicity was determined using the MTT assay 4 h after the completion of the irradiation. The results illustrated in Fig. 6 show that all samples displayed a low level of cytotoxicity in the dark, accounting for a good tolerance of the systems used. Besides, cells were not photosensitive in the absence of the photoactive compounds 1, 2 or 3. In contrast, considerable cell mortality was observed in the presence of these latter under illumination. In particular, the photoinduced mortality increased as a function of both the irradiation time and the concentration of the photoactive components. In all cases, the level of photodynamic inactivation induced by the conjugate 1 was higher as compared to the value observed with the model compounds 2 and 3 under the same experimental conditions. This finding provides clear-cut evidence for the involvement of a bimodal photo-inactivation mechanism in neoplastic destruction, in which NO and 1O2 are envisaged to play a key role. Note that, the amplified level of cell photomortality induced by the conjugate 1 is higher than that recently observed for the same cell lines in the case of a bichromophoric system in which the NO photodonor and the porphyrin centers were supramolecularly assembled in a ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.32 This is probably due to the photodelivery of both ROS and RNOS in the “very same region of space” of the cell compartment, as result of the covalent connection between the two photoprecursors in the conjugate 1.
:1.32 This is probably due to the photodelivery of both ROS and RNOS in the “very same region of space” of the cell compartment, as result of the covalent connection between the two photoprecursors in the conjugate 1.
 | ||
| Fig. 6 Dark and photoinduced mortality (λexc 400–800 nm) of melanoma cells incubated with dispersion of 4 loaded with 1, 2, or 3. | ||
Conclusions
We have developed herein a novel photoactivable molecular hybrid combining three-in-one photofunctionalities. The two chromogenic units of the conjugate “ignore” each other in the excited state, as proven by the excellent preservation of their photophysical and photochemical properties, and thus can be operated independently under the exclusive control of visible light inputs. Specifically, the porphryrin core exhibits satisfactory red fluorescence and excellent photosensitization of 1O2, while the nitroaniline acts as a NO photodispenser. Remarkably, this conjugate (i) can be delivered by polymer NPs in cancer cells, where it can be easily mapped by fluorescence microscopy and (ii) induces amplified level of cell mortality by bimodal action most likely due to a combined effect of the simultaneous photogeneration of ROS and RNOS in the same region of space. To our knowledge, this represents the first example of molecular hybrid exhibiting the convergence of dual therapeutic photoaction and imaging capability in a single molecular structure and opens fascinating possibilities for further studies on image-guided multimodal therapy in in vivo model systems.Experimental section
Synthesis
All reagents were of the highest commercial grade available and were used without further purification. All solvents used (from Sigma-Aldrich) were analytical grade. Compounds 1a and 3a were purchased from Sigma-Aldrich. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 at 300 and 75 MHz respectively, using SiMe4 as internal standard. Low resolution mass spectra were recorded with a Finnigan-Mat TSQ-700. High resolution mass spectra were recorded on a Bruker BioApex Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer equipped with an Apollo I ESI source, a 4.7 T superconducting magnet, and a cylindrical infinity cell (Bruker Daltonics, Billerica, MA, USA). Melting points were determined with a capillary apparatus (Büchi 540). Flash column chromatography was performed on silica gel (Merck Kieselgel 60, 230–400 mesh ASTM), or on neutral aluminum oxide (Fluka Aluminum Oxide for Chromatography, 0.05–0.15 mm, Brockmann activity I). The progress of the reactions was monitored by thin layer chromatography (TLC) on 5 cm × 20 cm plates with a layer thickness of 0.2 mm. Organic solvents were removed under vacuum at 30 °C. Elemental analyses (C, H, N) of the target compounds 1 and 3 were performed by REDOX Snc Monza, and the results are within 0.4% of the theoretical values. The purity of compound 2 (>97%) was assessed by HPLC. Analyses were performed on an Acquity Ultra Performance LC™, Waters Corporation Milford MA, USA, equipped with BSM, SM, CM and PDA detector. The analytical column was a Phenomenex Synergi 4 μ, Max-RP, 150 × 2 mm. Compound was dissolved in CH3OH. The mobile phase consisted of CH3OH/water with 0.1% formic acid 50/50. HPLC retention time (tR) was obtained at flow rates of 0.3 mL min−1, and the column effluent was monitored at 236 and 420 nm.The β-CD polymer 4 was prepared by crosslinking β-CD with epichlorohydrin, under strong alkaline conditions, following a previously described method.36
Sample preparation
Solution of 4 was prepared by stirring overnight 4 mg mL−1 of 4 in either aqueous phosphate buffer 10 mM at pH 7.4 or DMEM medium. Compounds 1, 2 and 3 were dissolved in methanol and slowly evaporated to form a thin film. These films were then hydrated with solutions of 1. The mixtures were stirred for 5 hours at 40 °C and then the final solutions were left to equilibrate at room temperature and filtered.Instrumentation
Steady-state absorption, emission, and NPs sizes. UV/Vis absorption and fluorescence spectra were recorded with a thermostated HP-8452 diode array spectrophotometer and Fluorolog-2 (Model, F-111) spectrofluorimeter respectively. All measurements were performed in a thermostated quartz cell (1 cm path length, 3 mL capacity). NPs sizes were measured by a dynamic light scattering Horiba LS 550 apparatus equipped with a diode laser with a wavelength of 650 nm.Fluorescence images were acquired using a Leica DMRB fluorescence microscope (Leica Microsystems Srl, Milan, Italy) equipped with a computer-assisted Nikon digital camera (Nital SpA, Turin, Italy).
| 4H+ + 2I− + 2NO2− → 2H2O + 2NO + I2 |
Irradiation was performed in a thermostated quartz cell (1 cm path length, 3 mL capacity, 25 °C) by using a 200 mW continuum laser with λexc = 405 nm. NO measurements were carried out under stirring with the electrode positioned outside the light path in order to avoid NO signal artefacts due to photoelectric interference on the ISO–NO electrode.
Determination of fluorescence, triplet and 1O2 quantum yields
Fluorescence quantum yields were determined by using optically matched solution at the excitation wavelength of compounds 1 and 2 and aqueous solution of para-tetrakis(-methylpyridiniumyl)porphyrin as a standard (Φf = 0.047)33 through the following equation:| Φf = Φf(s)(In2/I(s)n2(s)) |
Quantum yields for the triplet formation were determined by using optically matched solution at the excitation wavelength of compounds 1 and 2 and aqueous solution of the same standard as above (ΦT = 0.92).33 The top ΔA of the triplet signal from each sample was plotted as a function of the laser intensity. In this case the initial part of each set of data points is proportional to the product ΦT × εT–T, where ΦT and εT–T are the quantum yield of the triplet state and its molar absorption coefficient, respectively. By taking into account that all solutions are almost optically matched at the excitation wavelength and that large changes in the εT–T are fairly unlikely, being substantially unchanged the band profiles, ΦT values may be directly estimated by the different slopes (π) of the straight-lines obtained from the linear portion of the plots, via the simple equation;
| ΦT = ΦT(s)π/π(s) |
1O2 quantum yield were determined by using optically matched solution at the excitation wavelength of compounds 1 and 2 and 5,10,15,20-tetrakis(4-sulfonatophenyl)-21H,23H-porphyrin in D2O as a standard (ΦΔ = 0.6).43 The luminescence of the 1O2 was recorded at different intensity of the laser pulse and the luminescence at initial time (LΔ at t = 0) was extrapolated from the curve fitting. The values of LΔ at t = 0, were then plotted against the laser intensity, and the related slopes (χ) were compared. The values of ΦΔ were determined by using the following equation:
| ΦΔ = ΦΔ(s)χ/χ(s) |
Experiments with cells
American Type Culture Collection (Rockville, MD, USA) was maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS), 2.0 mM L-glutamine, 100 U/mL penicillin, 100 μg mL−1 streptomycin, and 25 μg mL−1 fungizone (Sigma-Aldrich, Italy), and incubated at 37 °C in humidified atmosphere containing 5% CO2. Cells from confluent cultures were detached using trypsin/EDTA and seeded in complete DMEM medium. For cell staining, the cells were cultured in 12-well culture dishes for 24 hours. The medium was removed and replaced with medium without phenol red containing the solution of the sample for 4 hours. The cells were first washed with PBS, then fixed with 4% formaldehyde. After washing with PBS cells were incubated with 4,6-diamino-2-phenylindole (DAPI) (1:10000; Invitrogen) for 10 min.
The photocytotoxicity experiments were carried out by irradiating the cells incubated either without or with the photoactive components with a 150 W Xe lamp through a cut-off filter at 400 nm. Cell proliferation was assessed by MTT assays, based on the conversion of a substrate containing a tetrazolium ring to spectrophotometrically detectable formazan by mitochondrial dehydrogenases. Briefly, cells were seeded at an initial density of 8 × 103 cells per microwell in flat-bottomed 200 μL microplates, incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 24 hours. Subsequently, part of the cells were maintained as media controls while others were incubated with the photoactive compounds. In both cases complete DMEM without phenol red was used. Next, 20 μL of 0.5% 3-(4,5-dimethyl-thiazol-2-yl)2,5-diphenyl-tetrazolium bromide in PBS were added to each microwell. Following 4 h of incubation at 37 °C, the supernatant was removed and replaced with 100 μL of DMSO. The optical density of each well sample was measured with a microplate spectrophotometer reader (Digital and Analog Systems, Rome, Italy) at 550 nm. The cell viability (%) was calculated according to the following equation:
| Cell viability (%) = [ABefore − (AAfter/ABefore)] × 100 |
Acknowledgements
We thank AIRC, Project IG-12834, for financial support to the AIRC fellow AF and funding of the research. Financial support from MIUR (PRIN 2011) and from the Marie Curie Program # 608407 CYCLON-HIT (FP7-PEOPLE-ITN-2013) is also acknowledged.Notes and references
- N. L. Komarova and C. R. Boland, Nature, 2013, 499, 291 CrossRef CAS PubMed.
- D. Lane, Nat. Biotechnol., 2006, 24, 163 CrossRef CAS PubMed.
- S. Sortino, J. Mater. Chem., 2012, 22, 301 RSC.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446 CrossRef CAS PubMed.
- Q. Shao and B. Xing, Chem. Soc. Rev., 2010, 39, 2835 RSC.
- B. Jang, J.-Y. Park, C.-H. Tung, I.-H. Kim and Y. Choi, ACS Nano, 2011, 5, 1086 CrossRef CAS PubMed.
- N. Stephanopoulos, G. J. Tong, S. C. Hsiao and M. B. Francis, ACS Nano, 2010, 4, 6014 CrossRef CAS PubMed.
- G. Szakács, J. K. Paterson, J. A. Ludwig, C. Booth-Gent and M. M. Gottesman, Nat. Rev. Drug Discovery, 2006, 3, 219 CrossRef PubMed.
- R. K. Pandey and G. Zheng, in The Porphyrin Handbook, ed. K. M. Smith, K. Kadish and R. Guilard, Academic Press, San Diego, 2000, vol. 6, pp. 157–230 Search PubMed.
- A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817 CrossRef CAS PubMed.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2004, 1, 279 CrossRef CAS.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Photodiagn. Photodyn. Ther., 2005, 2, 91 CrossRef CAS.
- Nitric Oxide: Biology and Pathobiology, ed. L. J. Ignarro, Elsevier Inc., 2010 Search PubMed.
- R. C. Jin and J. Loscalzo, J. Blood Med., 2010, 1, 147 CAS.
- J. V. Esplugues, Br. J. Pharmacol., 2002, 135, 1079 CrossRef CAS PubMed.
- J. E. Albina and J. S. Reichner, Cancer Metastasis Rev., 1998, 17, 39 CrossRef CAS.
- D. Fukumura, S. Kashiwagi and R. K. Jain, Nat. Rev. Cancer, 2006, 6, 521 CrossRef CAS PubMed.
- J. L. Wallace, Mem. Inst. Oswaldo Cruz, 2005, 100, 5 CAS.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091 CrossRef CAS PubMed.
- Nitric oxide donors, ed. P. G. Wang, T. B. Cai and N. Taniguchi, Wiley-VCH, Weinheim, 2005 Search PubMed.
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903 RSC.
- P. C. Ford, Nitric Oxide, 2013, 34, 56 CrossRef CAS PubMed.
- Q. Jia, A. J. Janczuk, T. Cai, M. Xian, Z. Wen and P. G. Wang, Expert Opin. Ther. Pat., 2002, 12, 819 CrossRef CAS.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 12, 2795 CrossRef PubMed.
- A. Fraix, N. Kandoth and S. Sortino, in Specialist Periodical Reports in Photochemistry, 2013, vol. 41, p. 302 Search PubMed.
- E. B. Caruso, S. Petralia, S. Conoci, S. Giuffrida and S. Sortino, J. Am. Chem. Soc., 2007, 129, 480 CrossRef CAS PubMed.
- S. Conoci, S. Petralia and S. Sortino, EP2051935A1/US20090191284, 2006.
- K. Fukuhara, M. Kurihara and N. Miyata, J. Am. Chem. Soc., 2001, 123, 8662 CrossRef CAS PubMed.
- D. K. Deda, C. Pavani, E. Caritá, M. S. Baptista, H. E. Toma and K. Araki, J. Porphyrins Phthalocyanines, 2012, 16, 56 CrossRef.
- E. B. Caruso, E. Cicciarella and S. Sortino, Chem. Commun., 2007, 5028 RSC.
- N. Kandoth, E. Vittorino, M. T. Sciortino, I. Colao, A. Mazzaglia and S. Sortino, Chem.–Eur. J., 2012, 18, 1684 CrossRef CAS PubMed.
- A. Fraix, R. A. Gonçalves, V. Cardile, A. C. E. Graziano, T. A. Theodossiou, K. Yannakopoulou and S. Sortino, Chem.–Asian J., 2013, 8, 2634 CrossRef CAS PubMed.
- K. Kalyanasundaram, Inorg. Chem., 1984, 23, 2453 CrossRef CAS.
- N. Kandoth, V. Kirejev, S. Monti, R. Gref, M. B. Ericson and S. Sortino, Biomacromolecules, 2014, 15, 1768 CrossRef CAS PubMed.
- S. Daoud-Mahammed, J. L. Grossiord, T. Bergua, C. Amiel, P. Couvreur and R. Gref, J. Biomed. Mater. Res., Part A, 2008, 86, 736 CrossRef CAS PubMed.
- M. Othman, K. Bouchemal, P. Couvreur, D. Desmaële, E. Morvan, T. Pouget and R. Gref, J. Colloid Interface Sci., 2011, 354, 517 CrossRef CAS PubMed.
- A. Fraix, N. Kandoth, I. Manet, V. Cardile, A. C. E. Graziano, R. Gref and S. Sortino, Chem. Commun., 2013, 49, 4459 RSC.
- E. Deniz, N. Kandoth, A. Fraix, V. Cardile, A. C. E. Graziano, D. Lo Furno, R. Gref, F. M. Raymo and S. Sortino, Chem.–Eur. J., 2012, 18, 15782 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
- S. Monti and S. Sortino, Chem. Soc. Rev., 2002, 31, 287 RSC.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113 CrossRef CAS PubMed.
- P. N. Coneski and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3753 RSC.
- J. Mosinger and Z. Micka, J. Photochem. Photobiol., A, 1997, 107, 77 CrossRef CAS.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |