
A convenient method for producing mono- and dichlorohydrins from glycerol
Donatella Giomi*a,
Marino Malavoltia,
Oreste Piccolob,
Antonella Salvini*a and
Alberto Brandia
aDipartimento di Chimica “Ugo Schiff”, Università di Firenze, Via della Lastruccia 3-13, 50019 Sesto Fiorentino (FI), Italy. E-mail: donatella.giomi@unifi.it; antonella.salvini@unifi.it; Tel: +39 055 457 3475 Tel: +39 055 457 3455
bSCSOP, Via Bornò 5, 23896 Sirtori, Italy
First published on 8th September 2014
Abstract
A new method for the transformation of glycerol into mono- and dichlorohydrins has been studied. With trimethylchlorosilane as chlorinating agent and acetic acid as catalyst, mono- and dichlorohydrins have been obtained in high yields and selectivity. In fact, under different reaction conditions, the synthesis of α-monochlorohydrin (3-chloropropan-1,2-diol) or α,γ-dichlorohydrin (1,3-dichloropropan-2-ol) as predominant product has been achieved. This process was also exploited for the valorisation of the crude mixture of glycerol and monochlorohydrin (glyceric mixture), a by-product from an earlier BioDiesel production. A reaction mechanism has been proposed based on investigations on the chlorination of different alcohols.
1. Introduction
Glycerol is a nontoxic renewable commodity, with many industrial applications in different domains (foods, beverages, cosmetics, paper, textiles, tobacco, etc.).1 Industrially, glycerol is obtained as a by-product by the saponification, hydrolysis or transesterification of triglycerides (animal fats and vegetable oils) during BioDiesel (BD) production. From these sources glycerol is recovered (ca. 10% by weight of the initial mass) in a crude state, and needs to be purified by distillation or ion exchange treatments. Other important pathways to produce glycerol are the synthesis from propene or by fermentation.1 Nowadays, the growing production of BD makes glycerol a cheap raw material for several derived products like ethers, esters, organic acids, acrolein and chlorohydrins.2–7 The chlorohydrins, in particular dichlorohydrins, are precursors of epichlorohydrin, an important intermediate in the production of epoxide resins and several fine chemicals.8–10For decades chlorohydrins have been made by hypochlorination of allyl chloride or by chlorination of allyl alcohol, but both these synthetic routes start from propene.8 Thus, chlorination of glycerol represents an interesting alternative, in particular starting from a renewable raw material originated from biomass.
The chlorination reaction of glycerol was already known at the beginning of the twentieth century. It was performed mixing glycerol with phosphorus trichloride or phosphorus pentachloride,11 by heating glycerol in the presence of disulfur chloride (S2Cl2)12,13 or thionyl chloride (SOCl2),14 or by hydrochloric acid (HCl) in the presence of small amounts of an organic acid (RCOOH).2,15–25 In the first three methods the stoichiometry of the reaction must be precisely controlled to avoid over-chlorination, while the last method gave high selectivity towards 3-chloropropan-1,2-diol (1-MCH or α-monochlorohydrin) and 1,3-dichloropropan-2-ol (1,3-DCH or α,γ-dichlorohydrin) with low production of 1,2,3-trichloropropane and other undesirable chlorinated ethers and oligomers.
The chlorination of glycerol with HCl/RCOOH system has been widely studied. Many different reaction conditions have been tested, including aqueous or anhydrous conditions, low or high pressure, use of pure or crude glycerol. Acetic acid has been the first applied catalyst15–19 and today it is still the cheaper one. However, a large number of other organic acids (as propionic, adipic, glutaric or malonic acid),2,20–25 their derivatives (like esters, amides, lactones, lactames)21 and even polymers (like polyacrylamide and polyacrylic acid)26 have been tested as catalysts. On the other hand, the chlorinating agent has been always hydrochloric acid (HCl), in water solution or in gas form. The optimization of the conversion and the selectivity of the reaction led to maximize the production of monochlorohydrin17,19,23 and, even more interestingly, to obtain dichlorohydrins, in particular 1,3-dichloropropan-2-ol in high yields.16,20–22 Many efforts have been performed also to propose a reaction mechanism.2,22,24,25,27,28
Since glycerol is a polyalcohol, a chlorinating agent selective for primary alcohol groups like (chloro-phenylthio-methylene)dimethylammonium chloride29 could be used to obtain 1-MCH and 1,3-DCH. However, in this case the price of the reactant is a limiting factor. Also trimethylchlorosilane (TMSCl) was found to be a good chlorinating agent for primary and tertiary alcohols.30 This last reaction occurs at room temperature if catalysed by dimethylsulfoxide (DMSO).
In literature, TMSCl has been also used as silylating agent of glycerol to obtain 1-trimethylsililoxypropan-2,3-diol31 and as chlorinating agent on epichlorohydrin for the synthesis of 1,3-DCH.32
In the present work we report on the use of TMSCl, in addition with a catalytic amount of acetic acid, to produce α,γ-dichlorohydrin or α-monochlorohydrin in good yields and selectivity, starting from glycerol. The application of TMSCl to the transesterification of tryglycerides33,34 for BioDiesel (BD) production has been recently studied in our laboratory. In this reaction two easily separable phases were obtained: the upper one consisted of fatty acid methyl esters useful for BD, while the other was a mixture of glycerol and α-monochlorohydrin in about equimolar ratio. In order to demonstrate the usefulness of the transesterification process in producing either BD or valuable derivatives of glycerol, the chlorination reaction in the present research has been performed also on this mixture of glycerol and α-monochlorohydrin.
Moreover hexamethyldisiloxane (HMDSO), the by-product coming from TMSCl, can be easily recovered from the reaction mixture and converted back into trimethylchlorosilane35 with HCl and zinc chloride, lowering the price of the process.
2. Experimental
All reactants have been used as purchased. Only trimethylchlorosilane has been distilled before use and purity (93% w/w) has been checked by 1H-NMR analysis. 13C-NMR spectroscopy has been used as semi-quantitative method to determine the molar ratio of the chlorinated products and the residual glycerol. NMR spectra have been recorded with Varian Mercury plus 400 and Varian VXR 200 instruments.2.1 Chlorination of glycerol
| Entry | TMSCl ratioa | AcOH ratioa | Temperature | Time | Distribution of products (molar ratio) by 13C-NMR | ||
|---|---|---|---|---|---|---|---|
| 1 (Gly) | 2 (1-MCH) | 3 (1,3-DCH) | |||||
| a Ratio between the moles of reagent and the moles of glycerol.b Catalyst: tartaric acid (see ref. 23).c Catalyst: DMSO (see ref. 30). | |||||||
| 1 | 2.9 | no | 100 °C | 24 h | 0.40 | 0.57 | 0.03 |
| 2 | 2.5 | 0.03 | 100 °C | 2 h | — | 0.63 | 0.37 |
| 3 | 2.9 | 0.04 | 100 °C | 6 h | — | 0.14 | 0.86 |
| 4 | 2.8 | 0.03 | 100 °C | 8 h | — | 0.12 | 0.88 |
| 5 | 2.8 | 0.03 | 100 °C | 12 h | — | 0.04 | 0.96 |
| 6 | 2.9 | 0.03 | 100 °C | 18 h | — | 0.01 | 0.99 |
| 7 | 2.5 | 0.04 | 60 °C | 6 h | 0.22 | 0.74 | 0.04 |
| 8 | 2.2 | 0.05 | 60 °C | 12 h | 0.05 | 0.77 | 0.18 |
| 9 | 2.5 | 0.04 | 60 °C | 18 h | — | 0.75 | 0.25 |
| 10 | 2.5 | 0.04 | 30 °C | 65 h | 0.29 | 0.68 | 0.03 |
| 11 | 1.1 | 0.03 | 100 °C | 18 h | 0.14 | 0.78 | 0.08 |
| 12 | 1.2 | 0.04 | 60 °C | 18 h | 0.16 | 0.78 | 0.05 |
| 13 | 1.9 | 0.04 | 60 °C | 12 h | 0.05 | 0.76 | 0.18 |
| 14 | 1.8 | 0.06 | 60 °C | 18 h | — | 0.68 | 0.32 |
| 15 | 2.5 | (0.08)b | 100 °C | 5 h | — | 0.47 | 0.53 |
| 16 | 3.0 | (0.47)c | 100 °C | 18 h | 0.19 | 0.81 | — |
2.2 Chlorination of the mixture of glycerol and monochlorohydrin (glyceric mixture) from BD production
Sunflower oil [19.0 g, 21.6 mmol for MW = 879 g mol−1], TMSCl (5.48 g, 6.5 ml, 46.9 mmol) and methanol (3.15 g, 4 ml, 98.0 mmol) were poured in a 50 ml flask. After heating for 8 hours at 60 °C under continuous stirring, the mixture has been left to separate in two phases. The upper one (fatty acid methyl esters for BD) was removed and the second phase was washed with petroleum ether and dried at room temperature under vacuum (10 mmHg) for 1 hour to eliminate the volatile compounds. The resulting mixture consisted in glycerol and α-monochlorohydrin (1.92 g, 10% w/w, molar ratio 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48 by 13C-NMR).
:48 by 13C-NMR).
| Entry | TMSCl ratioa | AcOH ratioa | Temperature | Time | Distribution of products (molar ratio) by 13C-NMR | ||
|---|---|---|---|---|---|---|---|
| 1 (Gly) | 2 (1-MCH) | 3 (1,3-DCH) | |||||
| a Ratio between the moles of reagent and the moles of hydroxy groups involved in each reaction.b Composition of the starting material. | |||||||
| Ab | 0.48 | 0.52 | |||||
| 1 | 2.7 | 0.05 | 100 °C | 12 h | — | 0.04 | 0.96 |
| 2 | 2.4 | 0.09 | 60 °C | 18 h | — | 0.29 | 0.71 |
| 3 | 2.7 | 0.07 | 60 °C | 9 h | 0.02 | 0.68 | 0.30 |
2.3 TMSCl chlorination of different alcohols and diols
| Entry | Alcohol | TMSCl ratioa | AcOH ratioa | Temperature | Time | Final molar ratio (1H-NMR or 13C-NMR)b | ||
|---|---|---|---|---|---|---|---|---|
| R–OH | R–Cl | R–OAc | ||||||
| a Ratio between the moles of reagent and the moles of alcohol.b Determined by comparison with literature data.c Traces of the ether derivative.d Mixture of 1-Cl-2-propanol (90%) and 2-Cl-1-propanol (10%). | ||||||||
| 1 | Benzyl alcohol | 1.1 | 0.98 | 60 °C | 9 h | — | 0.62 | 0.38 |
| 2 | Benzyl alcohol | 1.2 | 0.29 | 60 °C | 9 h | — | 0.85 | 0.15 |
| 3 | Benzyl alcohol | 1.2 | 0.06 | 60 °C | 12 h | — | 0.96 | 0.04 |
| 4 | n-Propyl alcohol | 1.2 | 0.28 | 60 °C | 9 h | 0.73 | — | 0.26 |
| 5 | n-Propyl alcohol | 1.2 | 0.06 | 60 °C | 12 h | 0.91 | 0.03 | 0.06 |
| 6 | n-Propyl alcohol | 1.2 | 0.06 | 100 °C | 15 h | — | 0.94 | 0.06c |
| 7 | Cyclohexanol | 1.3 | 0.27 | 60 °C | 9 h | 0.71 | — | 0.29 |
| 8 | Cyclohexanol | 1.2 | 0.27 | 100 °C | 15 h | — | 0.84 | 0.14c |
| 9 | Cyclohexanol | 1.2 | 0.06 | 100 °C | 15 h | 0.03 | 0.84 | 0.09c |
| 10 | t-Butyl alcohol | 1.2 | 0.27 | 60 °C | 9 h | — | 0.99 | — |
| 11 | t-Butyl alcohol | 1.2 | 0.07 | 60 °C | 12 h | — | 0.99 | — |
| 12 | 1,2-Propanediol | 2.3 | 0.06 | 60 °C | 12 h | 0.13 | 0.87d | — |
| 13 | Ethylene glycol | 2.2 | 0.06 | 60 °C | 12 h | — | 0.98 | 0.02 |
3. Results and discussion
The chlorination of glycerol (1) with hydrochloric acid, catalysed by an organic acid, is reported in Scheme 1. Depending on the reaction conditions (temperature, time, catalyst and quantity of HCl) monochlorination or polychlorination can be obtained. | ||
| Scheme 1 Chlorination of glycerol with HCl. | ||
As pointed out in previous works,2,20,21 the chlorination afforded predominantly 1-MCH (2) and 1,3-DCH (3) according with the higher reactivity and lower steric hindrance of primary alcohol groups with respect to secondary ones. Nevertheless small amount of 2-MCH (4), 1,2-DCH (5) and 1,2,3-trichloropropane (6) have been found, depending on the quantity of HCl used, temperature and reaction time. Moreover, the formation of minor chlorinated ethers and oligomers was also reported.20,21
The chlorination of glycerol with TMSCl is reported in Scheme 2. Surprisingly, among the 5 possible chloroderivatives of glycerol, only compounds 2 and 3 were obtained, also in the absence of catalyst (Table 1, entry 1).
 | ||
| Scheme 2 Chlorination of glycerol with TMSCl. | ||
The reaction was monitored by 13C-NMR spectroscopy as semiquantitative analysis technique. In fact, three CH signals, one for each molecule (glycerol, 3-chloropropan-1,2-diol and 1,3-dichloropropan-2-ol), are easily recognizable in the spectra (Fig. 1).
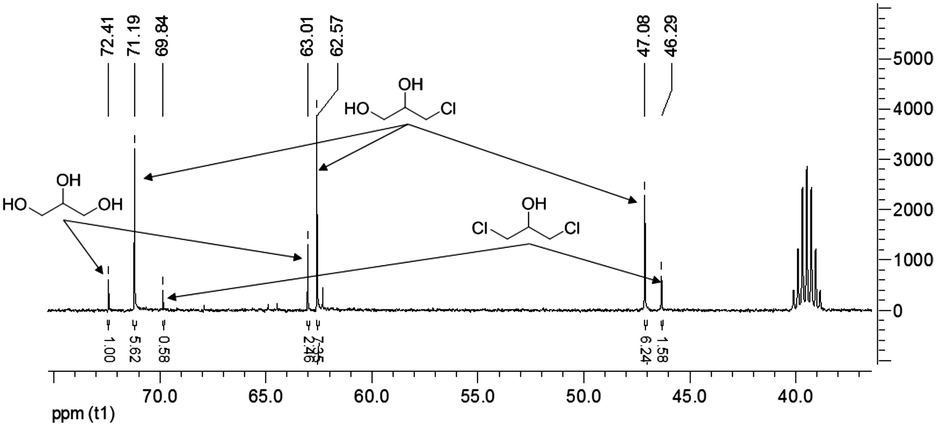 | ||
| Fig. 1 Typical analysis: estimation of glycerol and chloroderivatives content by 13C-NMR spectroscopy in DMSO-d6 as solvent. | ||
Several trials of chlorination of glycerol are reported in Table 1, performed by changing temperature and reaction times, and varying the amount of TMSCl and catalyst.
The presence of acetic acid was essential for the complete conversion of glycerol. In fact, without catalyst, after 24 hours at 100 °C glycerol was still present in the reaction mixture (Table 1, entry 1) while the conversion was complete with acetic acid (entry 2), in less than two hours at the same temperature.
By comparison of the experiments carried out at 100 °C (TMSCl/glycerol molar ratio range 2.5–2.9) (Table 1, entries 2–6, and Fig. 2) it can be observed that glycerol was totally converted in the first two hours of reaction in a 1.7:1 mixture of 1-MCH (2) and 1,3-DCH (3) (entry 2). As the chlorination proceeded, after six hours 3 is the main product (1:6 ratio, entry 3) and a conversion greater than 95% is reached after 12 hours (Table 1, entry 5).
 | ||
| Fig. 2 Experiments performed at 100 °C (Table 1, entries 2–6). | ||
As regards the experiments carried out at 60 °C (TMSCl/glycerol molar ratio in the range 2.2–2.5) (Table 1, entries 7–9, and Fig. 3), it can be observed that 1-MCH (2) was the main product with a predominance close to 80%. A similar 1-MCH (2) formation was also achieved by lowering the temperature to 30 °C and increasing the reaction time to 65 hours (Table 1, entry 10) or by reducing the amount of TMSCl (Table 1, entries 11–14). However, in these cases, if the molar ratio of TMSCl was slightly higher than 1 the glycerol conversion was not complete (entries 11 and 12) while, if the quantity of TMSCl was between 1.8 and 2.5, the conversion depended only on the time of reaction (compare entries 13 with 8 and 14 with 9).
 | ||
| Fig. 3 Experiments at 60 °C (Table 1, entries 7–9). | ||
Santacesaria and co-workers2,22–25 found a relation between the pKa of the organic acid used as catalyst and the degree of chlorination when HCl was used: acidic catalysts having pKa ≥ 4 were normally selective towards dichlorohydrin, while catalysts with pKa values in the range 1.2–3 were more selective for monochlorohydrins. In fact, also in the present chlorination procedure, we found a higher amount of 1-MCH (2) using tartaric acid (first pKa = 3.03) instead of acetic acid (pKa = 4.75) (Table 1, entries 3 and 15). However, the system TMSCl/tartaric acid resulted poorly selective (2 and 3 were obtained in almost equimolar ratio).
As final consideration, even if DMSO was reported30 as a good catalyst for chlorination of primary alcohols with TMSCl, in the case of glycerol the best results were obtained under AcOH catalysis (compare entries 6 and 16, Table 1).
The best reaction conditions to obtain selectively α-monochlorohydrin (2) or α,γ-dichlorohydrin (3) found in the experiments carried out on pure glycerol, were also used with the mixture of glycerol and α-monochlorohydrin, deriving from the transesterification of sunflower oil with TMSCl, and the results are reported in Table 2. The amounts of TMSCl and AcOH were referred to the hydroxy groups involved in each reaction. In particular, the stoichiometric ratio to obtain α,γ-dichlorohydrin (3) was 2 with respect to glycerol and 1 with respect to 1-MCH (2). The stoichiometric ratio to obtain α-monochlorohydrin (2) was 1 with respect to glycerol.
The 1,3-DCH (3) was obtained with high selectivity operating at 100 °C for 12 hours (Table 2, entry 1).
On the other hand, the formation of 2 as predominant product (2/3 molar ratio 68:30) was achieved by heating the starting mixture at 60 °C for 9 hours (Table 2, entry 3). Longer reaction times or higher temperatures strongly favour dichlorination (Table 2, entries 1 and 2).
Finally, in order to understand the type of mechanism involved in the reaction, the chlorination system TMSCl/AcOH was applied to several alcohols (Table 3). A survey of the literature showed that TMSCl is a good chlorinating agent for primary and tertiary alcohols when the reaction is catalyzed by DMSO at room temperature.30 The use of TMSCl with AcOH instead of DMSO, allows the chlorination of benzyl alcohol only by heating the reaction mixture at 60 °C. However, in our case the chlorination occurs also using a lower quantity of TMSCl, just above the stoichiometric ratio (1.2 equiv. vs. 2 equiv. used by Snyder30). It was found that, beside a complete conversion of the benzyl alcohol, the quantity of AcOH affects the selectivity of the reaction, because a side reaction affording benzyl acetate takes place (Table 3, entries 1–3). The best results were obtained with a molar ratio AcOH/ROH 0.06 by heating for 12 hours at 60 °C (Table 3, entry 3). A similar behaviour was also observed for t-butanol (Table 3, entries 10 and 11).
On the other hand n-propanol and cyclohexanol resulted much less reactive then benzyl alcohol and t-butanol in the same reaction conditions (Table 3, entries 4, 5 and 7). Only when heated at 100 °C for 15 hours they were converted into the corresponding chlorides (Table 3, entries 6, 8 and 9). It should be noted that secondary alcohols were found unreactive under the catalysis by DMSO.30
The reaction of 1,2-propanediol, using 2.3 equiv. of TMSCl, one equiv. for each hydroxyl group, showed a greater reactivity of this substrate with respect to the monofunctional alcohol (Table 3, entries 4–6 and 12), giving as main product 1-chloropropan-2-ol at lower temperature. Also ethylene glycol showed a great reactivity: in fact it was completely converted into the corresponding monochloro derivative.
The relevance of a possible generation of dry HCl by reaction of TMSCl with compounds having mobile protons (ROH or RCOOH) is, in our opinion, moderate. In analogy to Snyder,30 we assume that the mechanism of the reaction on monohydroxy compounds could involve the attack of the acid catalyst on TMSCl leading to adduct 9 that is now activated for the nucleophilic attack of the alcohol, with the release of the catalyst. The last step is a nucleophilic substitution of the chloride ion on the trimethylsilyl intermediate 10, leading to the chloro derivative (Scheme 3).
 | ||
| Scheme 3 Proposed mechanism for monofunctional alcohol chlorination. | ||
On the other hand, the great difference of reactivity observed for n-propanol and 1,2-propanediol suggests a different reaction mechanism for the two substrates. While mono hydroxyl substrates react through simple nucleophilic substitution pathways (Scheme 3), for polyols (1,2-propanediol, ethylene glycol and glycerol) a two-step process involving the formation of epoxide intermediates 12a–c and 13a–c could be proposed, according with some literature data†.2,22–25 In particular, the intermediate 13a (R = CH2OH) evolves into 1-MCH (2) by a nucleophilic attack of the chloride ion on the less hindered carbon atom (Scheme 4). The formation of the intermediates 12a–c from activated polyols 11a–c through intramolecular nucleophilic substitution can be described as an example of symphoria impossible for n-propanol and responsible for the higher reaction rate observed for 1,2-propanediol, ethylene glycol and glycerol. Furthermore, the presence of a 1,2-diol system on the α-monochlorohydrin (2) allows a second chlorination to give α,γ-dichlorohydrin (3).
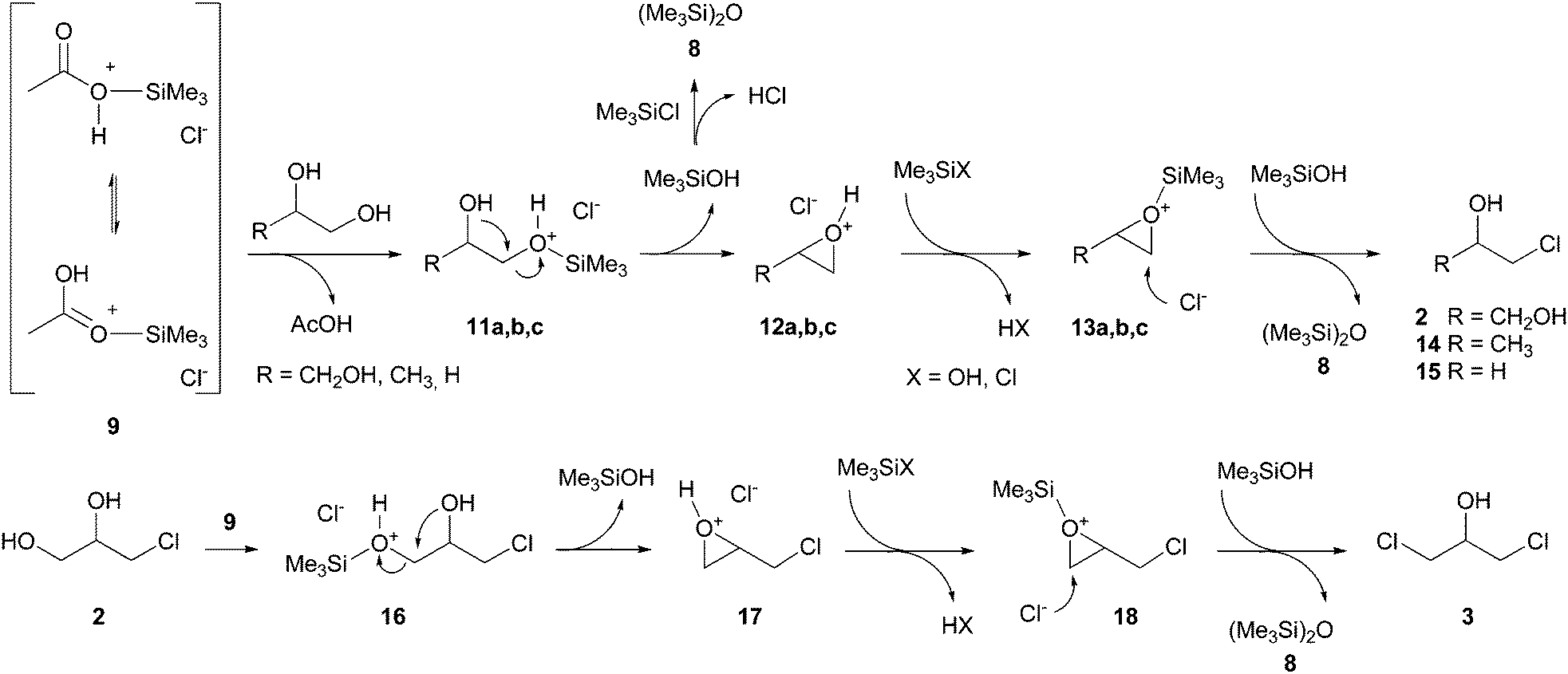 | ||
| Scheme 4 Proposed mechanism for chlorination of polyols. | ||
4. Conclusions
A new method for glycerol chlorination has been carried out using trimethylchlorosilane as chlorinating agent and AcOH as catalyst. The process has shown, under optimized conditions, a very high selectivity towards α,γ-dichlorohydrin (3), an useful starting material for the production of epichlorohydrin. By controlling the reaction conditions is also possible to drive the conversion of glycerol towards α-monochlorohydrin (2). Hexamethyldisiloxane (HMDSO) is formed as by-product of the reaction, that can be quantitatively recovered by distillation and conveniently transformed back to TMSCl,35 for recycling in the process. Moreover the present methodology could be easily integrated in the process for the transesterification of triglycerides with TMSCl,33,34 for the conversion of the mixture glycerol/monochlorohydrin, by-product of the BD production, into valuable α,γ-dichlorohydrin (3).Notes and references
- R. Christoph, B. Schmidt, U. Steinberner, W. Dilla and R. Karinen, Glycerol, in Ullmann's Encyclopedia of Industrial Chemistry, 2006 Search PubMed.
- E. Santacesaria, R. Vitiello, R. Tesser, V. Russo, R. Turco and M. Di Serio, Ind. Eng. Chem. Res., 2014, 53, 8939–8962 CrossRef CAS.
- X. Fan, R. Burton, Y. Zhou and others, Open Fuels Energy Sci. J., 2010, 3, 17–22 CrossRef CAS.
- F. Yang, M. A. Hanna and R. Sun, Biotechnol. Biofuels, 2012, 5, 1–10 CrossRef PubMed.
- R. De Sousa, C. Thurier, C. Len, Y. Pouilloux, J. Barrault and F. Jerome, Green Chem., 2011, 13, 1129–1132 RSC.
- C. Len and R. Luque, Sustainable Chemical Processes., 2014, 2, 1 CrossRef.
- H. Saggadi, D. Luart, N. Thiebault, I. Polaert, L. Estel and C. Len, RSC Adv., 2014, 4, 21456–21464 RSC.
- H. Q. Pham and M. J. Marks, Epoxy Resins, in Ullmann's Encyclopedia of Industrial Chemistry, 2005 Search PubMed.
- C. Len, A.-S. Boulogne-Merlot, D. Postel, G. Ronco, P. Villa, C. Goubert, E. Jeufrault, B. Mathon and H. Simon, J. Agric. Food Chem., 1996, 44, 2856–2858 CrossRef CAS.
- V. Legros, C. Vanhaverbeke, F. Souard, C. Len and J. Désiré, Eur. J. Org. Chem., 2013, 2583–2590 CrossRef CAS.
- M. Berthelot and S. De Luca, Il Nuovo Cimento, 1855, 2, 295–297 CrossRef.
- L. Carius, Justus Liebigs Ann. Chem., 1862, 122, 71–77 CrossRef.
- A. Claus, J. Chem. Soc., 1873, 26, 1117–1149 RSC.
- P. Carré and P. Mauclère, Compt. Rend., 1931, 192, 1567–1569 Search PubMed.
- Reboul and Lourenço, Justus Liebigs Ann. Chem., 1861, 119, 233–237 CrossRef.
- J. B. Conant and O. R. Quayle, Org. Synth., 1922, 2, 29 CrossRef.
- J. B. Conant and O. R. Quayle, Org. Synth., 1922, 2, 33 CrossRef.
- E. C. Britton and R. L. Heindel, Preparation of glycerol dichlorohydrin, US 2144612 A, 1939.
- E. C. Britton and H. R. Slagh, Manufacture of glycerol monochlorohydrin, US 2257899 A, 1941.
- P. Krafft, P. Gilbeau, B. Gosselin and S. Claessens, Process for producing dichloropropanol from glycerol, the glycerol coming eventually from the conversion of animal fats in the manufacture of biodiesel, WO 2005054167 A1, 2005.
- D. Schreck, W. J. Kruper, R. Varjian, M. E. Jones, R. Campbell, K. Kearns, B. D. Hook and J. G. Hippler, Conversion of a multihydroxylated-aliphatic hydrocarbon or ester thereof to a chlorohydrin, WO 2006020234 A1, 2006.
- R. Tesser, E. Santacesaria, M. Di Serio, G. Di Nuzzi and V. Fiandra, Ind. Eng. Chem. Res., 2007, 46, 6456–6465 CrossRef CAS.
- E. Santacesaria, M. Di Serio and R. Tesser, Process for monochlorohydrins production from glycerol and hydrochloric acid, WO 2008132770 A1, 2008.
- E. Santacesaria, R. Tesser, M. Di Serio, L. Casale and D. Verde, Ind. Eng. Chem. Res., 2010, 49, 964–970 CrossRef CAS.
- R. Tesser, M. Di Serio, R. Vitiello, V. Russo, E. Ranieri, E. Speranza and E. Santacesaria, Ind. Eng. Chem. Res., 2012, 51, 8768–8776 CrossRef CAS.
- A. Ten Kate, M. J. J. Mayer, C. J. G. Van Strien, B. Kuzmanovic and C. A. M. C. Dirix, Process for the catalytic halogenation of a hydroxylated organic compound, WO 2008074733 A1, 2008.
- T. P. Viswanathan and P. S. M. Kannan, Curr. Sci., 1978, 47, 802–803 CAS.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean: Soil, Air, Water, 2008, 36, 657–661 CrossRef CAS.
- L. Gomez, F. Gellibert, A. Wagner and others, Tetrahedron Lett., 2000, 41, 6049–6052 CrossRef CAS.
- D. C. Snyder, J. Org. Chem., 1995, 60, 2638–2639 CrossRef CAS.
- O. Mlejnek, Collect. Czech. Chem. Commun., 1969, 34, 1777–1785 CrossRef CAS.
- L.-W. Xu, L. Li, C.-G. Xia and P.-Q. Zhao, Tetrahedron Lett., 2004, 45, 2435–2438 CrossRef CAS PubMed.
- A. Brandi, A. Salvini, G. Cipriani, D. Giomi and G. Bartolozzi, Process for the production of biodiesel by transesterification of triglycerides, WO 2011023712 A1, 2011.
- A. Salvini, D. Giomi, G. Cipriani, G. Bartolozzi, R. Alfini and A. Brandi, RSC Adv., 2012, 2, 4864–4868 RSC.
- B. Zhang and L. Shi, Hebei Gongye Keji, 2007, 24, 63–65 Search PubMed.
Footnote |
| † Formation of a cyclic intermediate coming from acetylated polyols could be also proposed, see ref. 28 and R. Boschan and S. Winstein, J. Am. Chem. Soc., 1956, 78, 4921–4925. |
| This journal is © The Royal Society of Chemistry 2014 |