
Aliphatic/aromatic spacers based azo dye dimers: synthesis and application for optical storage devices†
Abstract
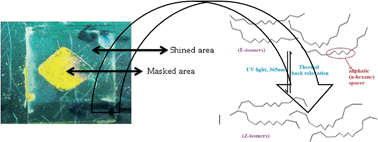
The photoisomerization effect of new bent-shaped azo dyes in the presence of aliphatic and aromatic spacers is reported for the first time. The synthesized compounds with n-hexane and benzene as central moieties were characterized by different spectral analytical techniques such as 1H-NMR, 13C-NMR, FTIR and UV-Vis. They revealed the photoisomerization effect in solution and on the solid cells as well. In solution, the E–Z and Z–E isomerization occurred in around 17–18 seconds and 7–13 hours, respectively. The dramatic variation in back relaxation is speculated to take place due to the nature of the spacers involved in the system. The synthesized materials are expected to be more useful in optical storage devices.
 Please wait while we load your content...
Please wait while we load your content...

