
Continuous flow synthesis of β-amino acids from α-amino acids via Arndt–Eistert homologation†
Vagner D.
Pinho
,
Bernhard
Gutmann
and
C. Oliver
Kappe
*
Institute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at
First published on 13th August 2014
Abstract
A fully continuous four step process for the preparation of β-amino acids from their corresponding α-amino acids utilizing the Arndt–Eistert homologation approach is described.
Less abundant than α-amino acids, β-amino acids represent an important class of bioactive compounds.1,2 The presence of an α methylene in β-amino acids gives β-amino acids higher conformational flexibility compared to their α-amino analogues, strongly affecting the structural and biological properties of β-peptides, such as secondary structure and the rate of enzymatic degradation.1,2 Many secondary metabolites found in nature have β-amino acids incorporated in their structure, and the biological activities of these compounds are frequently connected to the β-amino acid moiety, which makes them potential lead structures for the development of new drugs.1
Despite the plethora of interesting approaches for the preparation of β-amino acids,1,3 the use of readily available N-protected α-amino acids as starting materials for an Arndt–Eistert homologation sequence4b is particularly attractive, since the stereogenic center of the α-amino acids is retained in the homologated product without significant racemization (Scheme 1).1,2c,3b,4,5c The last step in this sequence, the Wolff rearrangement,5 can be performed thermally under silver catalysis, or photochemically induced,5b,c the latter approach being environmentally more friendly and suitable to continuous flow processing.6,7 Photochemical transformations in continuous flow have several advantages compared to their batch counterparts, since common problems associated with photochemistry, such as scalability and over irradiation can be overcome.6 The relatively small channel diameters and the high surface-to-volume ratio of capillary flow reactors assure efficient and uniform irradiation of the reaction mixture for a well-defined time, controlled by the pump flow rate.6
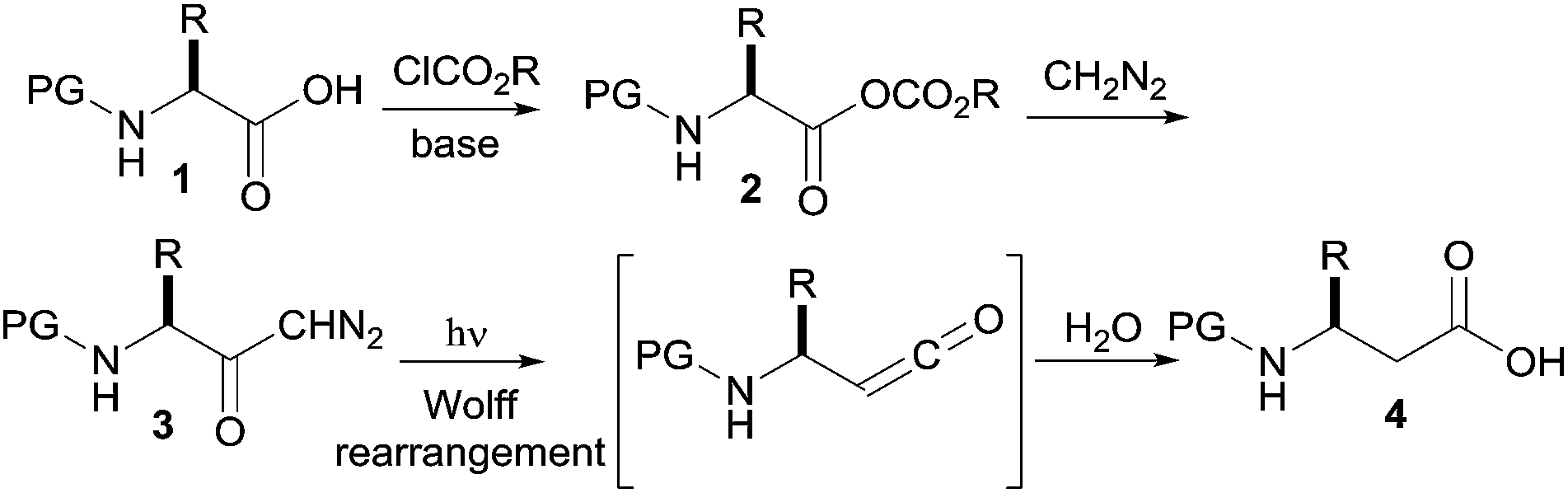 | ||
| Scheme 1 Preparation of β-amino from α-amino acids. | ||
The major drawback of the Arndt–Eistert homologation sequence is related to safety concerns associate with the use of diazomethane (CH2N2), a highly volatile, irritating, poisonous and carcinogenic compound. Furthermore, diazomethane is exceedingly heat-, light- and shock sensitive, and tends to decompose explosively.8 Recently, our group and others have developed strategies where generation, extraction and consumption of diazomethane were integrated in a continuous flow systems.9,10 Key to these protocols was the use of a semi-permeable, micro-porous, hydrophobic membrane, which selectively allows gases but not liquids to cross, to accomplish the selective separation of anhydrous CH2N2 from an aqueous feed.9 This approach was recently used by our group for the preparation of α-halo ketones, chiral building blocks for the synthesis of HIV protease inhibitors.9b
Herein we report a fully continuous four step synthesis of β -amino acids from the respective protected α -amino acids. For the synthesis of β -amino acids 4, four successive reactions have to be accomplished (Scheme 1): activation of the amino acid 1 to the mixed anhydride 2, acylation of diazomethane by the mixed anhydride 2 to form the α-diazoketone 3, and finally the Wolff rearrangement of the diazoketone with accompanying interception of the intermediate ketene with water to form the β -amino acids 4.
To achieve the continuous four-step synthesis of β-amino acids, we envisaged to couple the continuous flow preparation of α-diazoketones9 with a subsequent photochemically induced Wolff rearrangement. The initial optimization of the continuous photochemical Wolff rearrangement was performed with the pure α-diazoketone derived from phenylalanine (3a). Several compact fluorescent lights (CFL) were tested as light sources,6,11 and different reactor configurations and residence times were investigated (for details see the ESI†). The final photochemical reactor consisted of a commercial germicide compact fluorescent UV light (254 nm) surrounded by 3 mL of an UV-transparent perfluoroalkoxy alkane (PFA) tubing with an outer diameter of 1.5 mm. The heat generated by the lamp was dissipated from the reactor by air cooling with a fan. To ensure a stable flow rate, a back-pressure regulator (BPR, 6.9 bar) was attached at the downstream side of the photoreactor. For the flow reaction, a 0.16 M solution of the diazoketone 3a in THF was combined with a second feed of H2O/THF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in a T-mixer and the combined stream passed through the photoreactor. Complete conversion of the diazo compound 3a was obtained with flow rates of 150 μL min−1 for both feeds (residence time in the photo reactor: 10 min) and the pure β-amino acid 4a was isolated in 57% yield by column chromatography.
:1 in a T-mixer and the combined stream passed through the photoreactor. Complete conversion of the diazo compound 3a was obtained with flow rates of 150 μL min−1 for both feeds (residence time in the photo reactor: 10 min) and the pure β-amino acid 4a was isolated in 57% yield by column chromatography.
With the optimized conditions for the photochemical step in hand, we then evaluated the fully continuous synthesis of β-amino acids starting directly from α-amino acids. Therefore, the diazoketone was synthesized as reported previously from the respective activated α-amino acid in a commercially available tube-in-tube (TiT) reactor.9,12 The inner tube of that device is made of a hydrophobic, gas-permeable Teflon AF-2400 membrane (0.8 mm inner diameter, 1 mm outer diameter, 4 m length).12 For the generation of the anhydrous diazomethane, a 0.6 M methanolic solution of N-methyl-N-nitroso-p-toluenesulfonamide (Diazald) and a 1.2 M solution of KOH in MeOH/H2O 1:1 were pumped at flow rates of 200 μL min−1 into a T-mixer by two syringe pumps. The combined stream passed through a short residence tube and then further through the inner tube of the TiT reactor (Fig. 1). Diazomethane is formed in the inner tube and subsequently diffuses through the hydrophobic, permeable wall into the outer chamber. The activation of the N-protected α-amino acid was accomplished by means of a 1 mL residence loop (RT1), a T-mixer (TM1), two injection valves (SL1, SL2) and two further syringe pumps attached upstream to the outer tube of the TiT reactor (Fig. 1). A 2 mL solution of N-cbz-L-phenylalanine (0.64 mmol) and tributylamine (1.0 equivalent; feed C) in dry THF and a 2 mL solution of ethyl chloroformate (0.96 mmol, 1.5 equivalents; feed D) in dry THF were loaded into the sample loops of the two injection valves. When the flow reaction was started, the injection valves were switched to connect the sample loop in line with the carrier stream (dry THF). The two reaction solutions were pumped with flow rates of 75 μL min−1 into the T-mixer (TM1) and the merged stream then went through the tube reactor RT1 at room temperature. The mixture subsequently passed through the outer chamber of the TiT reactor where the activated acid reacted with the dry diazomethane generated in the inner tube (Fig. 1).9 Diazoketone formation was completed in a second residence loop (RT2) attached downstream to the outer tube of the tube-in-tube reactor. Finally, a solution of THF/H2O 1:1 was fed into the system at a flow rate of 150 μL min−1 using a fifth pump and a further T-piece (TM3). The reaction stream then went through the photoreactor at a combined theoretical flow rate of 300 μL min−1 (Fig. 1). Formation of the diazoketone from the activated acid liberates one equivalent of an acid and thus sequesters one equivalent of CH2N2 with an accompanying release of N2 gas. Furthermore, due to photochemical and/or thermal decomposition of excessive CH2N2 and diazoketone downstream of the TiT reactor, a considerable volume of nitrogen was generated. Therefore, the reaction mixture was driven through the photoreactor at considerably higher flow rates, resulting in relatively short residence times and incomplete rearrangement of the diazoketone. Moreover, the presence of excess of diazomethane during the photochemical Wolf rearrangement could cause side-reactions and could adversely affect the purity of the photochemical reaction step. Thus, in order to remove the generated nitrogen and any excess of diazomethane a 2 mL tubing of gas-permeable Teflon AF-2400 was attached between the RT2 and TM3 units (RT3; Fig. 1).13 The permeable tubing was immersed in an alcoholic solution of acetic acid to immediately quench any diazomethane diffused through it. This approach was successful in degassing the stream coming from the RT2, providing a stable and diazomethane-free flow stream to the photoreactor. The β-amino acid 4a was finally collected and isolated by chromatography in 40% product yield after the continuous flow four-step reaction sequence. The continuous flow setup was then used to prepare a variety of β-amino acids from the corresponding N-protected α-amino acids in yields comparable to those reported for conventional batch procedures (Fig. 2).14
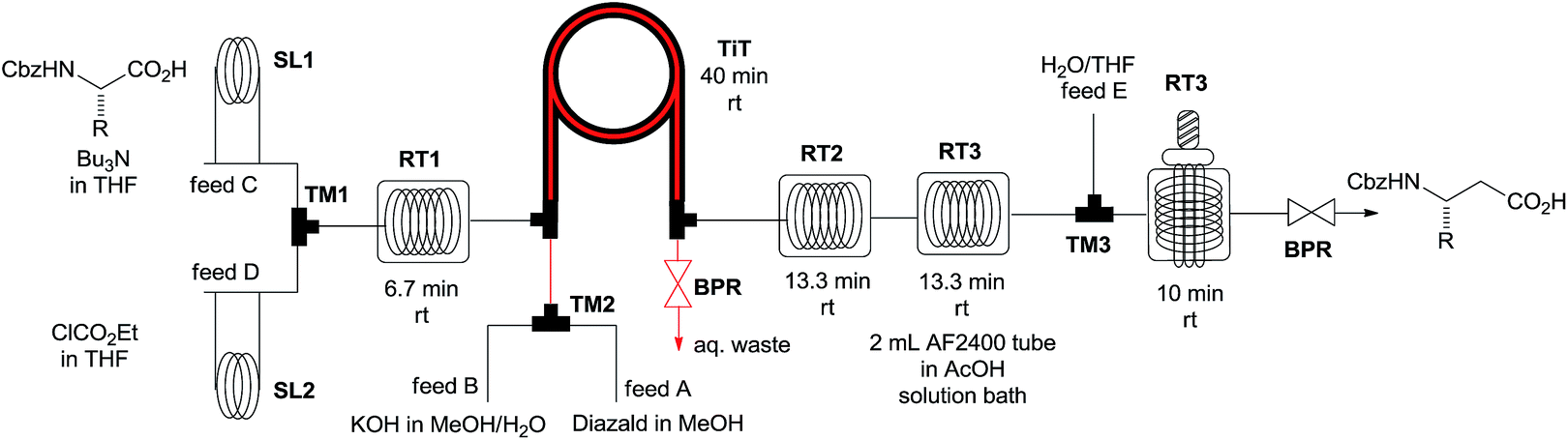 | ||
| Fig. 1 Flow set-up for the continuous four-step Arndt–Eistert homologation of α-amino acids. | ||
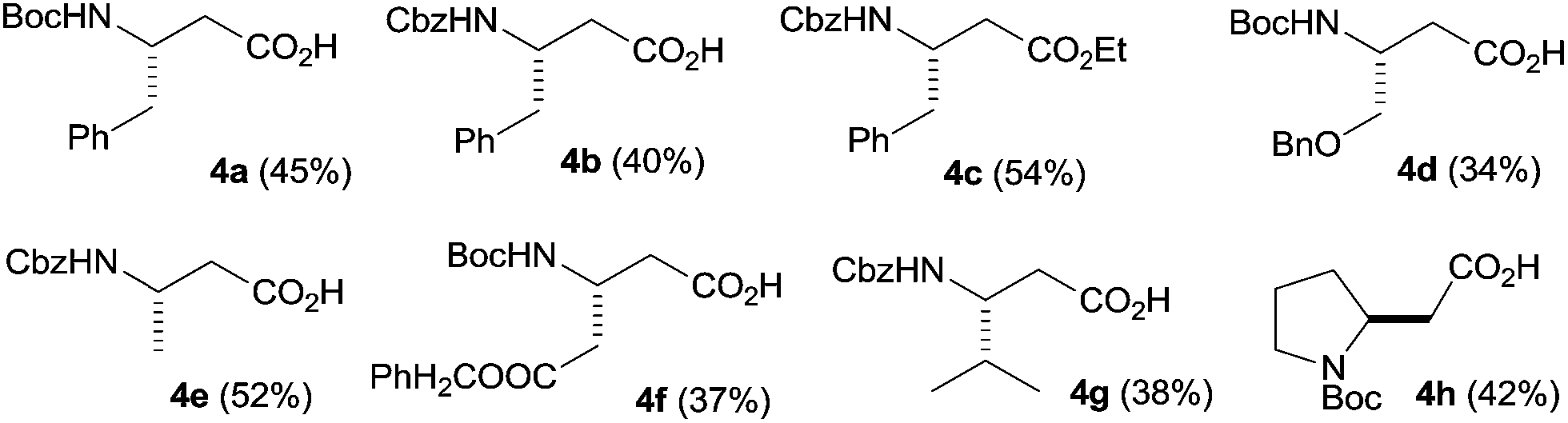 | ||
| Fig. 2 β-amino acids prepared in continuous flow (Fig. 1). | ||
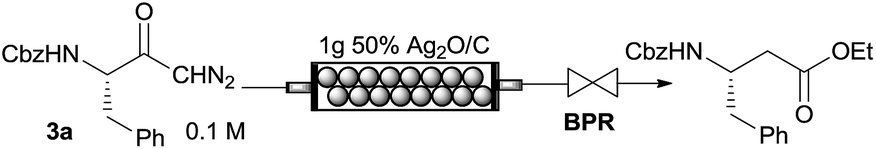
As an alternative to the photochemical Wolff rearrangement, we briefly also explored the silver-catalyzed rearrangement using reaction cartridges packed with silver oxide.5c,15 Accordingly, 2 mL of a 0.16 M solution of diazoketone 3a in THF/EtOH 1:1 was pumped through a column packed with 1 g of silver oxide/charcoal 1:1 (Table 1). Applying a flow rate of 400 μL min−1 at room temperature a conversion of 76% to the homologated ester was obtained. Full conversion was achieved with a residence time of ∼10 min at a reaction temperature of 60 °C. The pure homologated product was isolated in 71% yield by chromatography (Table 1). ICPMS analysis of the processed samples indicated that the catalyst is highly stable and does not leach under the investigated reaction conditions (∼0.05 ppm Ag were detected in the samples; see ESI†). These preliminary results suggest the participation of a heterogeneous silver species in the catalytic cycle. Further investigations on mechanistic aspects of the silver catalyzed rearrangement are ongoing in our laboratories.
Conclusions
In conclusion, we have demonstrated a fully continuous four-step synthesis of β-amino acids from α-amino acids following the Arndt–Eistert homologation approach. For this process the diazomethane was generated on-demand in a microreactor environment and directly extracted from the aqueous feed using a gas-permeable membrane. The anhydrous CH2N2 thus generated was consumed by acylation with the activated amino acid. Any excess of CH2N2 was immediately removed from the reaction stream employing a second gas-selective membrane and destroyed in a quench solution. A subsequent photo-Wolff rearrangement provided the β-amino acids in reasonable overall yields. The on-demand generation–purification–consumption of diazomethane, as described herein, eliminates all human exposure to this hazardous reagent and reduces the risk of explosive decomposition. The use of membranes to separate and purify hazardous gases may allow a plethora of other synthetic chemistry to be explored and performed in the future.Acknowledgements
V.D.P. thanks CAPES for a postdoctoral scholarship (PGCI 5502-13-6).Notes and references
- For a discussion of the synthesis and biology of β -amino acids see: in Juaristi and V. A. Soloshonok, Enantioselective Synthesis of Beta-Amino Acids, Wiley-VCH, Hoboken, N.J., 2nd edn 2005 Search PubMed.
- (a) S. Abele, K. Vögtli and D. Seebach, Helv. Chim. Acta, 1999, 82, 1539 CrossRef CAS; (b) D. Seebach, K. Gademann, J. V. Schreiber, J. L. Matthews, T. Hintermann, B. Jaun, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1997, 80, 2033 CrossRef CAS; (c) D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 13 Search PubMed; (d) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed.
- (a) B. Weiner, W. Szymański, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656 RSC; (b) M. Liu and M. P. Sibi, Tetrahedron, 2002, 58, 7991 CrossRef CAS; (c) D. C. Cole, Tetrahedron, 1994, 50, 9517 CrossRef CAS; (d) N. Sewald, Angew. Chem., Int. Ed., 2003, 42, 5794 CrossRef CAS PubMed; (e) E. Juaristi, D. Quintana and J. Escalante, Aldrichimica Acta, 1994, 27, 4397 Search PubMed; (f) A. Liljeblad and L. T. Kanerva, Tetrahedron, 2006, 62, 5831 CrossRef CAS PubMed; (g) L.-W. Xu and C.-G. Xia, Eur. J. Org. Chem., 2005, 633 CrossRef CAS; (h) C. J. Saavedra, A. Boto and R. Hernández, Org. Biomol. Chem., 2012, 10, 4448 RSC.
- (a) F. Arndt, B. Eistert and W. Partale, Ber. Dtsch. Chem. Ges., 1927, 60, 1364 CrossRef; (b) J. Podlech and D. Seebach, Angew. Chem., Int. Ed., 1995, 34, 471 CrossRef CAS; (c) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS.
- (a) L. Wolff, Justus Liebigs Ann. Chem., 1902, 325, 129 CrossRef; (b) H. Meier and K. Zeller, Angew. Chem., Int. Ed., 1975, 14, 32 CrossRef; (c) W. Kirmse, Eur. J. Org. Chem., 2002, 2002, 2193 CrossRef.
- For reviews on flow photochemistry, see: (a) J. P. Knowles, L. D. Elliot and K. I. Booker-Milburn, Beilstein J. Org. Chem., 2012, 8, 2025 CrossRef CAS PubMed; (b) T. Noel, Chim. Oggi, 2013, 31, 10 CAS; (c) M. Oelgemöller, Chem. Eng. Technol., 2012, 35, 1144 CrossRef; (d) K. Gilmore and P. H. Seeberger, Chem. Rec., 2014, 14, 410 CrossRef CAS PubMed; (e) E. E. Coyle and M. Oelgemöller, Photochem. Photobiol. Sci., 2008, 7, 1313 RSC.
- For examples of photochemical Wolff rearrangements in continuous flow, see: (a) Y. S. M. Vaske, M. E. Mahoney, J. P. Konopelski, D. L. Rogow and W. J. McDonald, J. Am. Chem. Soc., 2010, 132, 11379 CrossRef CAS PubMed; (b) T. P. Willumstad, O. Haze, X. Y. Mak, T. Y. Lam, Y.-P. Wang and R. L. Danheiser, J. Org. Chem., 2013, 78, 11450 CrossRef CAS PubMed; (c) S. Garbarino, L. Banfi, R. Riva and A. Basso, J. Org. Chem., 2014, 79, 3615 CrossRef CAS PubMed.
- T. H. Black, Aldrichimica Acta, 1983, 16, 3 CAS.
- (a) F. Mastronardi, B. Gutmann and C. O. Kappe, Org. Lett., 2013, 15, 5590 CrossRef CAS PubMed; (b) V. D. Pinho, B. Gutmann, L. S. M. Miranda, R. O. M. A. de Souza and C. O. Kappe, J. Org. Chem., 2014, 79, 1555 CrossRef CAS PubMed.
- (a) R. A. Maurya, C. P. Park, J. H. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2011, 50, 5952 CrossRef CAS PubMed; (b) P. Poechlauer, S. Braune, B. Dielemans, B. Kapein, R. Obermüller and M. Thathagar, Chim. Oggi, 2012, 30, 51 CAS.
- For a recent example of using CFLs in flow reactors from our laboratory, see: D. Cantillo, O. de Frutos, J. A. Rincón, C. Mateos and C. O. Kappe, Org. Lett., 2014, 16, 896 CrossRef CAS PubMed.
- The tube-in-tube device was originally developed in the Ley laboratory as a gas-addition tool: M. O'Brien, I. R. Baxendale and S. V. Ley, Org. Lett., 2010, 12, 1596 CrossRef PubMed.
- For the use of gas-permeable Teflon AF-2400 for gas-removing purposes see: (a) K. Skowerski, S. J. Czarnocki and P. Knapkiewicz, ChemSusChem, 2014, 7, 536 CrossRef CAS PubMed; (b) P. Koos, U. Gross, A. Polyzos, M. O'Brien, I. Baxendale and S. V. Ley, Org. Biomol. Chem., 2011, 9, 6903 RSC; (c) M. O'Brien, N. Taylor, A. Polyzos, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 1250 RSC; (d) M. J. Fink, M. Schön, F. Rudroff, M. Schnürch and M. D. Mihovilovic, ChemCatChem, 2013, 5, 724 CrossRef CAS.
- M. R. Linder, S. Steurer and J. Podlech, Org. Synth., 2002, 79, 154 CrossRef CAS.
- M. S. Newman and P. F. Beal, J. Am. Chem. Soc., 1950, 72, 5163 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, reaction optimization, and characterization of all compounds. See DOI: 10.1039/c4ra08113g |
| This journal is © The Royal Society of Chemistry 2014 |