
Catalytic transfer hydrogenation of levulinate esters to γ-valerolactone over supported ruthenium hydroxide catalysts†
Yasutaka Kuwahara*ab,
Wako Kaburagia and
Tadahiro Fujitania
aResearch Institute for Innovation in Sustainable Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
bDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: kuwahara@mat.eng.osaka-u.ac.jp; Fax: +81-6-6105-5029; Tel: +81-6-6879-7458
First published on 16th September 2014
Abstract
Production of γ-valerolactone (GVL) from levulinate esters and several alcohols as hydrogen donors via a catalytic transfer hydrogenation (CTH) process was performed over supported ruthenium hydroxide catalysts. Among the catalysts examined, Ru(OH)x supported on high-surface-area, anatase TiO2 containing highly-dispersed ultrasmall Ru(OH)x nano-clusters was found to be the most active catalyst, which converted levulinate esters to GVL in an almost quantitative yield under mild reaction conditions with low catalyst loading. Addition of heterogeneous bases could afford a faster reaction rate in GVL production by accelerating the following intramolecular dealcoholation step. The catalyst was reusable over repeated catalytic cycles without loss of catalytic performance, making this material a potential candidate for efficient GVL production under ambient reaction conditions.
Introduction
With increasing concern about the strong dependence on petroleum and the associated adverse effects on the environment, worldwide efforts have recently been devoted to development of advanced technologies to produce fuels and chemicals from biomass resources, which is a so-called “biomass refinery”. The key reason of this trend is that biomass is an abundant, renewable and carbon-neutral source, and is available worldwide. A number of studies in this area have demonstrated that lignocellulosic biomass has a great potential to be converted into fuels and a variety of chemicals.1–7 Of various chemicals synthesized from lignocellulosic biomass, γ-valerolactone (GVL) has been regarded as one of the most valuable molecules,8 since it can be used as a fuel additive,9 solvent for biomass processing,10 as well as an ideal precursor for the production of alkanes,11,12 alkenes,13–15 and other valuable chemicals.16,17 Current route to produce GVL from cellulosic biomass includes (i) the hydrogenation (using molecular H2) or (ii) the catalytic transfer hydrogenation (CTH) (using alcohols) of levulinic acid (LA) and its esters,18 which are intermediate compounds produced by acid-catalyzed solvolysis of various carbohydrate fractions of lignocelluloses, such as cellulose, glucose, fructose, as well as 5-hydroxymethylfurfural (HMF), in water19–21 and alcohols,22–27 respectively (Scheme 1). The former hydrogenation reaction is generally conducted by using various metal catalysts such as Ru,28–33 Pd,33–35 Pt,33 Ir,36 Cu,37,38 however the systems require high-pressure H2 (>30 bar). The latter CTH process is an alternative viable option for scalable GVL production, since the reaction proceeds under milder conditions without the need of flammable, high-pressure H2 and alcohols are used as inexpensive and recyclable H-donors.39 Moreover, in an actual process, the production of levulinate esters from carbohydrates in alcohol media allows higher product yields and easier product separation than the production of LA in water,22,40 and the spent alcohol media can potentially be reused as H-donors in the following CTH process. Therefore, levulinate esters are better alternative intermediates for the energy-saving, cost-effective manufacture process of GVL.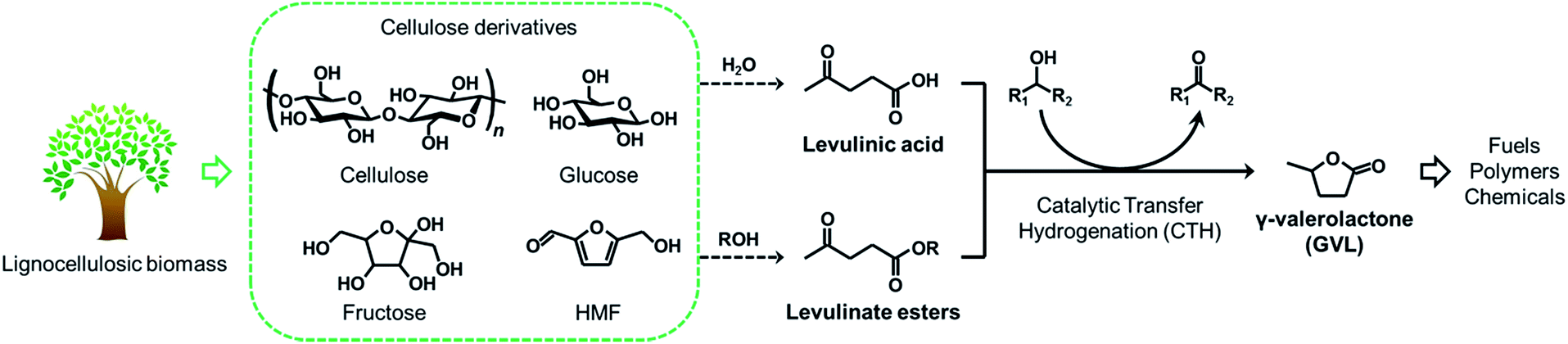 | ||
| Scheme 1 Production route of γ-valerolactone (GVL) from lignocellulosic biomass. | ||
Several catalytic systems have recently been reported for the production of GVL via a CTH process. Dumesic et al. reported a CTH process to convert LA and its esters into GVL using ZrO2 catalyst and isopropanol/isobutanol as H-donors at 150 °C under pressurized inert gas flow.41 Analogously, Sun and Lin et al. investigated catalytic activity of hydrated zirconia, ZrO(OH)2·xH2O, for a CTH reaction of ethyl levulinate in a supercritical ethanol.42,43 Microporous zirconosilicate molecular sieves (Zr-beta) has recently been reported to be more active and stable catalysts compared with bulk zirconia.44,45 However, the series of these precedent works have demonstrated that the CTH process over Zr-based catalysts suffer from drawbacks such as high catalyst loading, relatively high reaction temperature/pressure, moderate GVL selectivity and appreciable catalyst deactivation during the reaction. Apart from these, Guo and Fu et al. reported a room-temperature CTH reaction using Raney Ni preactivated in 2-propanol solution;46 however, the catalyst is sensitive to air and a long-time storage of catalyst in alcohol media is needed, which are critical drawbacks in a practical large-scale synthesis of GVL. Therefore, development of an alternative catalytic system to efficiently produce GVL from levulinate esters under ambient reaction conditions is still a matter of concern.
Herein, we report a CTH reaction of levulinate esters to produce GVL over ruthenium hydroxides supported on basic oxides under ambient reaction conditions (90 °C, atmospheric pressure) and with low catalyst loading (0.8 mol%). Ru is the cheapest metal among noble metals, and its hydroxide form is expected to act as an efficient heterogeneous catalyst for this reaction owing to its approved catalytic ability in the CTH reaction of carbonyl compounds.47,48 In the present catalytic system, GVL was produced through sequential CTH and dealcoholation reactions, being catalyzed by ruthenium hydroxide and basic sites of oxides, respectively. In particular, highly-dispersed ruthenium hydroxide species supported on high-surface-area, anatase TiO2 showed a high catalytic activity for this reaction. The catalyst could afford an almost quantitative yield of GVL under the optimal conditions and was readily reusable over multiple cycles without loss of catalytic performances.
Results and discussion
GVL production via a CTH process
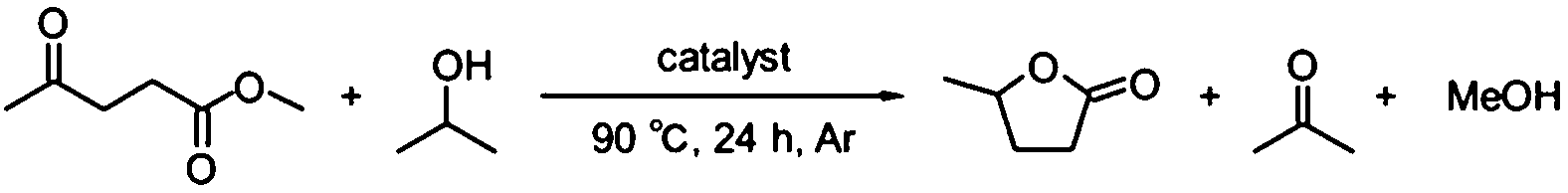
In the initial studies, Ru(OH)x catalysts supported on a variety of metal oxides were prepared and tested in the CTH reaction of methyl levulinate (ML) using 2-PrOH as a hydrogen donor. The X-ray diffraction (XRD) patterns of supported ruthenium hydroxide catalysts were identical to those of the parent supports and no diffraction peaks assignable to Ru(OH)x and RuO2 were observed in all cases (see Fig. S1 in ESI†), suggesting that ruthenium species are highly dispersed on the support surface. Table 1 summarizes the catalytic results together with Ru loading and the surface area determined by the BET (Brunauer–Emmett–Teller) method from N2 adsorption isotherms. The BET surface areas of the supported ruthenium hydroxide catalysts were close to those of the parent supports in all cases.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ru loading (wt%) | SBETb (m2 g−1) | Conversionc (%) | GVL yieldc (%) |
| a Reaction conditions: catalyst (Ru 0.8 mol%), methyl levulinate (1 mmol), 2-PrOH (5 mL), 90 °C, 24 h, Ar.b Determined by BET method.c Determined by GC using biphenyl as the internal standard. | |||||
| 1 | Ru(OH)x/TiO2(A) | 0.83 | 336 | 86 | 76 |
| 2 | Ru(OH)x/TiO2(AR) | 0.82 | 53 | >99 | 80 |
| 3 | Ru(OH)x/TiO2(R) | 0.84 | 46 | >99 | 49 |
| 4 | Ru(OH)x/α-Al2O3 | 0.84 | 6.7 | 78 | 38 |
| 5 | Ru(OH)x/γ-Al2O3 | 0.84 | 236 | 70 | 64 |
| 6 | Ru(OH)x/ZrO2 | 0.83 | 70 | 52 | 48 |
| 7 | Ru(OH)x/CeO2 | 0.83 | 130 | 1.2 | 0.6 |
| 8 | Ru(OH)x/MgO | 0.85 | 346 | 1.3 | 0.8 |
| 9 | Ru(OH)x/HT | 0.85 | 87 | 50 | 43 |
| 10 | Ru(OH)x | — | 63 | 52 | 47 |
| 11 | RuO2 | — | n.d. | No reaction | |
| 12 | Ru metal | — | n.d. | No reaction | |
| 13 | TiO2(A) | — | 339 | No reaction | |
Ruthenium hydroxide (Ru(OH)x) as a baseline sample afforded 47% GVL yield in 24 h of reaction time (entry 10), while neither RuO2 nor Ru metal showed activity in this reaction (entries 11 and 12). When the same amount of Ru was used, Ru(OH)x supported on TiO2 particularly gave higher ML conversions compared to the unsupported Ru(OH)x and the other supported catalysts even under mild reaction conditions with only 0.8 mol% of the catalysts. In this reaction conditions using 2-PrOH as a H-donor, byproducts associated with transesterification (e.g., isopropyl levulinate), that are frequently identified over bulk zirconia catalysts, were hardly observed.41 An important aspect is given from Ru(OH)x supported on different Al2O3 supports; Ru(OH)x supported on γ-Al2O3 gave a superior GVL yield (64%) than that supported on α-Al2O3 (38%) in 24 h of reaction, although both provided similar conversions of ML (70–78%) (cf. entries 4 and 5). Ru(OH)x/α-Al2O3 mainly afforded 4-hydroxy pentanoic acid methyl ester (4-HPM), which is a reaction intermediate to be transformed to GVL via an intramolecular dealcoholation. This difference is probably due to different number of basic site, i.e., abundant basic sites present on γ-Al2O3 (0.122 mmol g−1) more efficiently promote the dealcoholation step of 4-HPM to produce GVL than those on α-Al2O3 (0.023 mmol g−1) (for CO2-TPD data, see Fig. S2 in ESI†). Nevertheless, Ru(OH)x supported on typical basic supports such as MgO and CeO2 were almost inactive for this reaction (entries 7 and 8), and those supported on ZrO2 and Mg–Al hydrotalcite (HT) showed activities almost comparable to that of unsupported Ru(OH)x (entries 6 and 9), suggesting that the catalytic activity is also influenced by crystalline structure of support materials.
Among the catalysts tested, Ru(OH)x catalysts supported on TiO2 provided the highest conversions of ML, which far outperformed that given by unsupported Ru(OH)x. Among three different types of TiO2 supports (anatase TiO2 (TiO2(A), ST-01, Ishihara Sangyo Co., Ltd., SBET = 339 m2 g−1), anatase and rutile TiO2 (TiO2(AR), P25, AEROSIL Japan Industries, SBET = 55 m2 g−1), and rutile TiO2 (TiO2(R), TTO-55, Ishihara Sangyo Co., Ltd., SBET = 47 m2 g−1)), TiO2 possessing anatase crystalline phase gave apparently higher GVL yields (76–80%, entries 1–2) compared to pure rutile TiO2 (49%, entry 3). This is also attributed to the presence of larger amount of basic sites on anatase TiO2 crystals (0.091–0.119 mmol g−1) than that on rutile TiO2 crystals (0.057 mmol g−1), which can promote the intramolecular dealcoholation step (see Fig. S2 in ESI†).
Next, the effects of Ru loading on the catalytic activity were studied by altering the weight percentage of Ru metals on the catalysts (0.8, 2 and 4 wt% Ru). The XRD patterns remained unchanged upon increasing the Ru loadings (see Fig. S1 in ESI†). Fig. 1 compares ML conversion and GVL yield at 3 h of reaction on Ru(OH)x/TiO2(A) and Ru(OH)x/TiO2(AR) with varied Ru loadings. When TiO2(AR) (SBET = 55 m2 g−1) was used as a support, the activity monotonically decreased as Ru loading increased from 0.82 to 4.2 wt%. In contrast, Ru(OH)x supported on high-surface-area TiO2(A) (SBET = 339 m2 g−1) showed a continual activity increase as increasing the Ru loading, and 4% Ru(OH)x/TiO2(A) afforded the highest activity (89% yield of GVL with >99% conversion at 90 °C after 24 h of reaction; for detailed reaction kinetics, see Fig. 5). This result indicates a strong activity dependence on surface area of TiO2 supports.
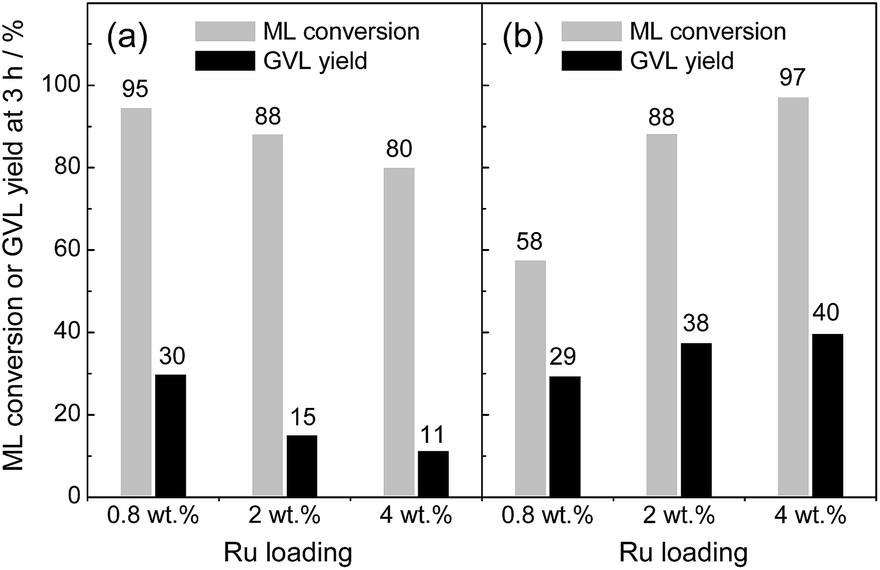 | ||
| Fig. 1 Comparison of methyl levulinate (ML) conversion and GVL yield at 3 h of reaction on (a) Ru(OH)x/TiO2(AR) and (b) Ru(OH)x/TiO2(A) with different Ru loadings. Reaction conditions: catalyst (Ru 0.8 mol%), ML (1 mmol), 2-PrOH (5 mL), 90 °C, 3 h, Ar. | ||
XAFS analysis of Ru(OH)x/TiO2
To clarify the local structure of ruthenium species on the TiO2 supports, X-ray absorption fine structure (XAFS) measurement was carried out. Fig. 2 shows Ru K-edge X-ray absorption near edge structure (XANES) spectra and radial distribution functions (RDF) obtained from the k3-weighted extended XAFS (EXAFS) oscillations of Ru(OH)x/TiO2(AR) together with those of Ru(OH)x as a reference sample. It is worth noting that pure Ru(OH)x has one-dimensional chain-like structure consisting of RuO6 octahedral units connected by two oxygen atoms with each other.48 The Ru K-edge XANES spectra of Ru(OH)x/TiO2(AR) are almost identical to that of Ru(OH)x, showing that the Ru atoms are likely present on TiO2(AR) surface in ruthenium(III) hydroxide form (Fig. 2(A)). In the RDF, two distinct peaks were observed at around r = 1.6 and 2.7 Å (the phase shift uncorrected), which correspond to Ru–O and Ru⋯Ru bonds, respectively (Fig. 2(B)). From curve-fitting analysis, the coordination number (C.N.) of the first Ru–O shell was determined to be about six in all cases (Table 2), again proving that Ru atoms have octahedral coordination similar to that of Ru(OH)x. However, the intensity of the second Ru⋯Ru shell signals was obviously weakened compared to that of pure Ru(OH)x. Curve-fitting analysis of this signal showed that the interatomic distance between two neighboring Ru atoms was unchanged (3.11 ± 0.01 Å), but the C.N. was varied from 0.91 to 1.1 by increasing the Ru loading from 0.82 to 4.2 wt% (Table 2), suggesting the generation of more aggregated Ru(OH)x species at higher Ru loadings. With the increase in the C.N.s, the catalytic activity monotonically decreased, indicating that highly-dispersed ruthenium hydroxide species is the main active sites for the present CTH reaction.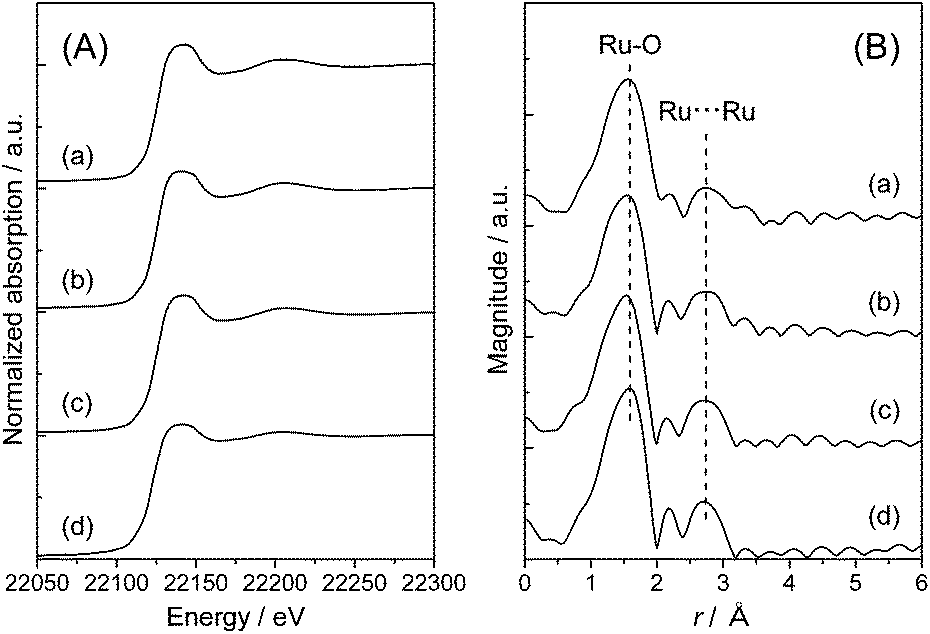 | ||
| Fig. 2 (A) Ru K-edge XANES spectra and (B) Ru K-edge radial distribution functions of Ru(OH)x/TiO2(AR) catalysts with varied Ru content ((a) 0.8 wt%, (b) 2 wt% and (c) 4 wt%) and (d) Ru(OH)x. | ||
| Catalyst | Ru loading (wt%) | SBETb (m2 g−1) | Shell | Rc (Å) | C.N.d |
|---|---|---|---|---|---|
| a k3-Weighted EAXFS, 3.0 ≤ k (Å−1) ≤ 11.0.b Determined by BET method.c Average interatomic distance.d Average coordination number. | |||||
| 0.8% Ru(OH)x/TiO2(AR) | 0.82 | 53 | Ru–O | 2.02 | 6.2 |
| Ru⋯Ru | 3.10 | 0.91 | |||
| 2% Ru(OH)x/TiO2(AR) | 2.1 | 59 | Ru–O | 2.02 | 6.1 |
| Ru⋯Ru | 3.12 | 1.0 | |||
| 4% Ru(OH)x/TiO2(AR) | 4.2 | 61 | Ru–O | 2.01 | 6.1 |
| Ru⋯Ru | 3.11 | 1.1 | |||
| 0.8% Ru(OH)x/TiO2(A) | 0.83 | 336 | Ru–O | 2.02 | 5.6 |
| Ru⋯Ru | 2.86 | 0.44 | |||
| 2% Ru(OH)x/TiO2(A) | 2.1 | 349 | Ru–O | 2.02 | 5.6 |
| Ru⋯Ru | 2.97 | 0.68 | |||
| 4% Ru(OH)x/TiO2(A) | 4.0 | 214 | Ru–O | 2.01 | 5.5 |
| Ru⋯Ru | 3.00 | 0.76 | |||
| Ru(OH)x | — | 63 | Ru–O | 2.02 | 6.3 |
| Ru⋯Ru | 3.11 | 1.2 | |||
A similar analysis was also performed on Ru(OH)x/TiO2(A). The XANES spectra of Ru(OH)x/TiO2(A) catalysts were very similar to that of Ru(OH)x at all Ru loading levels (Fig. 3(A)), indicating the formation of ruthenium(III) hydroxide species on anatase TiO2 crystals. In the RDF, the C.N. for the Ru–O shell at r = 1.6 was estimated to be approximately six; however, the intensity of the Ru⋯Ru shell signals were considerably weaker compared to pure Ru(OH)x or even that supported on TiO2(AR), and a shrinkage of Ru⋯Ru interatomic distance was observed (Fig. 3(B)). Curve-fitting analysis of the Ru⋯Ru shell signals showed that the C.N. is varied from 0.44, 0.68 and 0.76 by increasing the Ru loading to 0.83, 2.1 and 4.0 wt%, respectively. These values are apparently lower than those observed on Ru(OH)x/TiO2(AR) catalysts. Transmission electron microscope (TEM) image of 4% Ru(OH)x/TiO2(A) exhibited Ru clusters with the size of less than 3 nm throughout the particle surface (see Fig. S3 in ESI†). These combined analyses indicate that Ru species on TiO2(A) surface are present mainly as highly-dispersed ultrasmall ruthenium hydroxide nano-clusters and that a high-surface-area TiO2 can offer a surface environment suitable for generating such a ruthenium hydroxide species. Although the cause of the increased catalytic activity on Ru(OH)x/TiO2(A) cannot be fully understood from this aspect (other configurational factors and the balance between Ru(OH)x sites and support oxides may also affect simultaneously), it appears certain that highly-dispersed ultrasmall ruthenium hydroxide nano-clusters act as the main active sites for the present CTH reaction.
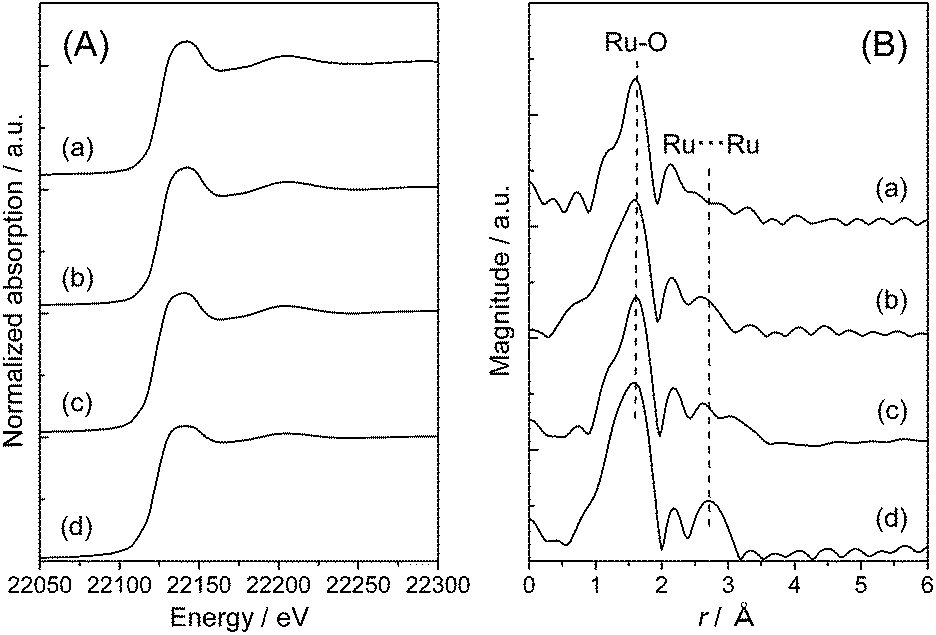 | ||
| Fig. 3 (A) Ru K-edge XANES spectra and (B) Ru K-edge radial distribution functions of Ru(OH)x/TiO2(A) catalysts with varied Ru content ((a) 0.8 wt%, (b) 2 wt% and (c) 4 wt%) and (d) Ru(OH)x. | ||
Effect of base additives
The reaction kinetics in the GVL production from ML and 2-propanol over 4% Ru(OH)x/TiO2(A), the best catalyst among examined, is shown in Fig. 4 (left, top), which reveals that the present GVL production route involves sequential two reaction steps; (i) a CTH reaction to reduce ML to 4-HPM by Ru(OH)x catalyst and (ii) the following intramolecular dealcoholation reaction of 4-HPM to produce GVL by basic sites, where the latter step dominates the GVL production rates. To obtain a faster GVL production rate by accelerating the dealcoholation step, addition of bases was also examined.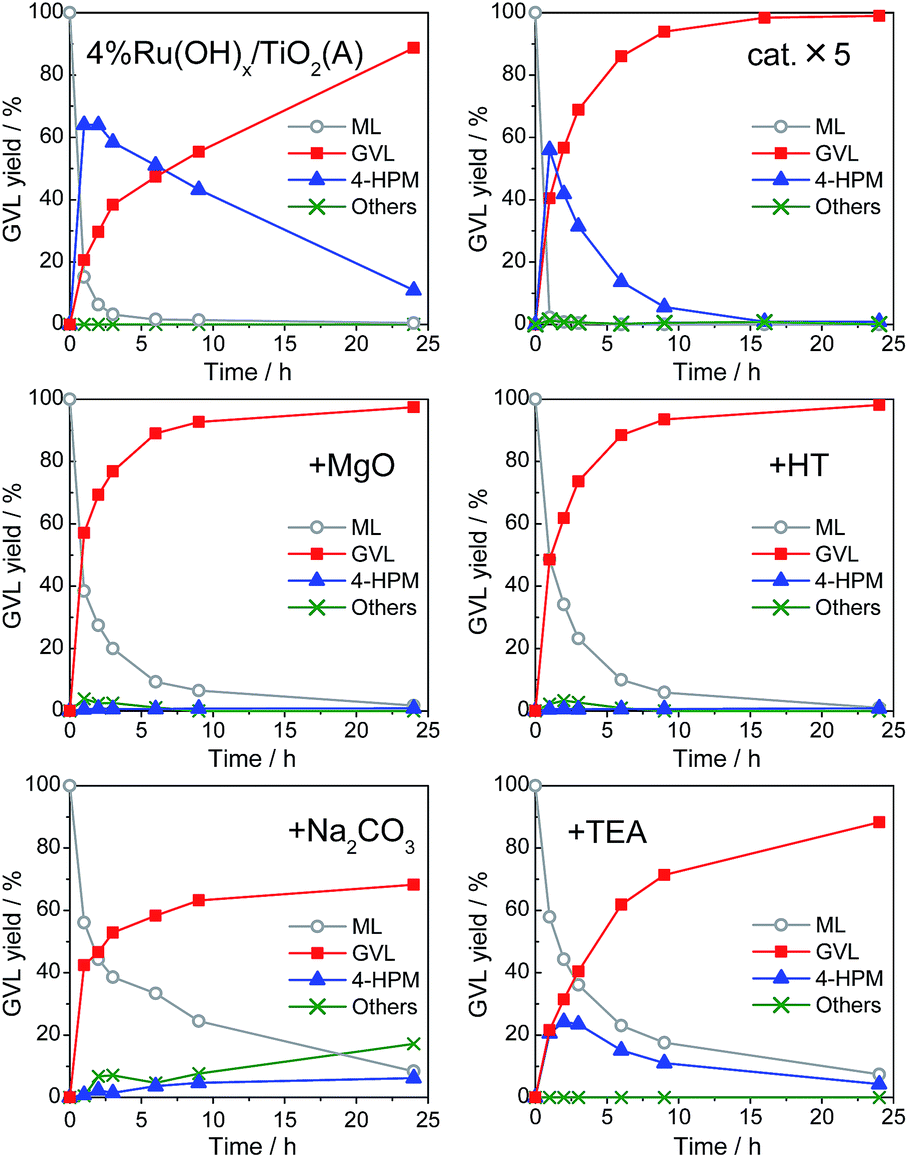 | ||
| Fig. 4 Effect of base additives in GVL production from ML with 2-PrOH over 4% Ru(OH)x/TiO2(A) catalyst. HT = hydrotalcite, TEA = triethylamine. Reaction conditions: catalyst (Ru 0.8 mol%), ML (1 mmol), 2-PrOH (5 mL), 90 °C, additive (1 mmol or 100 mg), Ar. Others may contain byproducts associated with transesterification reaction. | ||
Comparison of reaction kinetics in Fig. 4 clearly reveals that heterogeneous bases such as MgO and hydrotalcite (HT) act as effective co-catalysts, affording GVL in almost quantitative yields (>98%) within 24 h of reaction (Fig. 4, middle), whereas the ruthenium hydroxides supported on MgO and HT showed subpar catalytic activities for this reaction. The GVL production rates were as fast as that given with 4.0 mol% Ru catalyst (Fig. 4, right top), demonstrating the efficacy of base addition. Under the presence of MgO or HT, the formation of 4-HPM was significantly suppressed and GVL was dominantly produced. This is due to the prominent basicity of the heterogeneous bases to effectively promote the dealcoholation step of 4-HPM to form GVL (see Fig. S2 in ESI†). On the other hand, addition of typical homogeneous bases such as Na2CO3 and triethylamine (TEA) scarcely affected GVL yield, but rather reduced the conversion rate of ML, although the formation of 4-HPM was somehow suppressed (Fig. 4, bottom). They are likely to interact directly with Ru(OH)x sites and interrupt the CTH reaction due to their homogeneities. In particular, in the presence of Na2CO3, appreciable amounts of byproducts associated with transesterification reaction were formed owing to its strong basicity.
Scope of substrates and alcohols
Using the optimized catalyst, 4% Ru(OH)x/TiO2(A), we examined the scope of substrates and alcohols in the CTH process (Table 3). When ML was used as a starting material, other secondary alcohols such as 2-BuOH and cyclohexanol also efficiently afforded GVL with moderate yields (entries 5 and 6), but 2-PrOH afforded the highest GVL yield under the same reaction conditions (for reaction kinetics, see Fig. S4 in ESI†). In situ FTIR measurement with a flow of 2-propanol vapor in He at 90 °C confirmed the generation of covalently-bound iso-propoxyl groups, indicative of dissociative adsorption of 2-propanol over 4% Ru(OH)x/TiO2(A) catalyst (for in situ FTIR difference spectra, see Fig. S5 in ESI†). In the present CTH reaction, the thus formed alkoxy species are considered to easily undergo the β-H elimination, and two H-atoms remained on the Ru site are considered to readily react with ML to give 4-HPM via the formation of 6-ring transition state.46,49 On the other hand, primary alcohols such as methanol, ethanol and 1-propanol did not act as efficient H-donors under the reaction conditions examined in this study (entries 1–3). This is due to the difficulty in β-hydride elimination from primary alcohols.46
|
||||
|---|---|---|---|---|
| Entry | Substrate | Alcohol | Conversionc (%) | GVL yieldc (%) |
| a Reaction conditions: catalyst (Ru 0.8 mol%, 20 mg), substrate (1 mmol), alcohol (5 mL), 90 °C, 24 h, Ar.b At 60 °C.c Determined by GC.d Ru 4.0 mol% (100 mg). | ||||
| 1b | ML | MeOH | 0 | 0 |
| 2 | ML | EtOH | 2.2 | 0.8 |
| 3 | ML | 1-PrOH | 2.0 | 0.6 |
| 4 | ML | 2-PrOH | >99 (100)d | 89 (99)d |
| 5 | ML | 2-BuOH | 84 | 67 |
| 6 | ML | CyOH | 57 | 54 |
| 7 | EL | 2-PrOH | >99 (100)d | 71 (88)d |
| 8 | BL | 2-PrOH | >99 (100)d | 63 (81)d |
| 9 | LA | 2-PrOH | 10.8 (80)d | 3.5 (64)d |
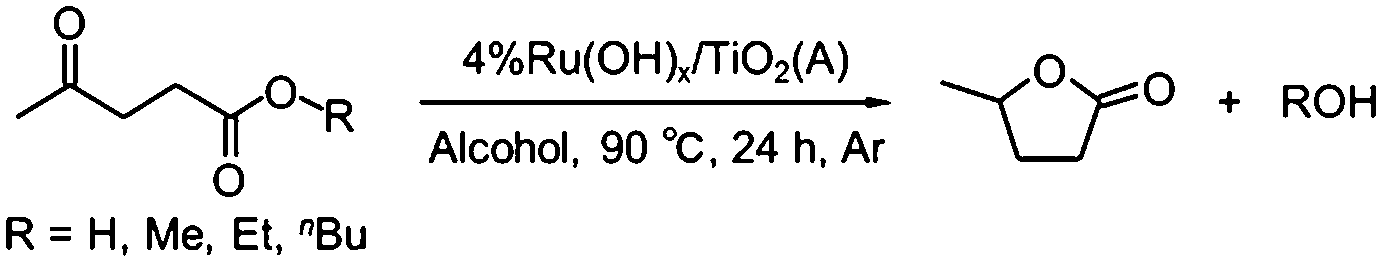
GVL was successfully synthesized from ethyl levulinate (EL) and butyl levulinate (BL) in good to excellent yields (entries 7 and 8); the reaction of EL and BL at 90 °C for 24 h with 0.8 mol% Ru resulted in 71% and 63% yields of GVL with >99% conversions, respectively. With 4.0 mol% Ru catalyst, EL and BL afforded 88% and 81% GVL yields, respectively. In both cases, the reaction proceeded with the same manner as that of ML, i.e., levulinate esters underwent a CTH step to produce the corresponding 4-hydroxypentanoates, followed by intramolecular dealcoholation to form GVL. As for the reaction of LA, 64% GVL yield was attained after 24 h of reaction with 4.0 mol% Ru catalyst, while GVL was scarcely produced from LA when 0.8 mol% Ru catalyst was used (entry 9). This is primarily due to the strong adsorption of acidic LA on basic Ru(OH)x sites and possibly due to the water molecules generated during the reaction. The reaction rate decreased in the order of: ML > EL > BL ≫ LA (for reaction kinetics, see Fig. S3 in ESI†), showing that the present catalytic system is especially effective for the transformation of levulinate esters to GVL.
Recyclability of catalyst
Since the lifetime of the catalysts has vital effect on the cost of the overall process, reusability and stability of the catalysts are of great importance for practical applications. To evaluate the reusability and stability of the catalysts, catalyst recycle tests for the CTH reaction of ML with 2-PrOH were performed over four successive cycles using 4% Ru(OH)x/TiO2(A) catalyst. After each catalytic run, the catalyst was separated from the reaction mixture by filtration, washed with acetone and water, dried under vacuum, and then subjected to next catalytic run. As shown in Fig. 5, the prepared 4% Ru(OH)x/TiO2 catalyst was at least four times reusable without loss of catalytic activity and selectivity (89 and 99% GVL yields after 24 h of reaction with 0.8 and 4.0 mol% Ru catalyst, respectively, even in the 4th use). ICP analysis of the solution after the reaction showed that elution of Ru species is negligible. XAFS analysis of the spent catalyst confirmed that the configuration of Ru species was unchanged upon the reaction (Fig. S6 in ESI†). These results show that Ru(OH)x/TiO2(A) is a highly effective and recyclable heterogeneous catalyst for the CTH reaction of levulinate esters to give GVL under mild reaction conditions.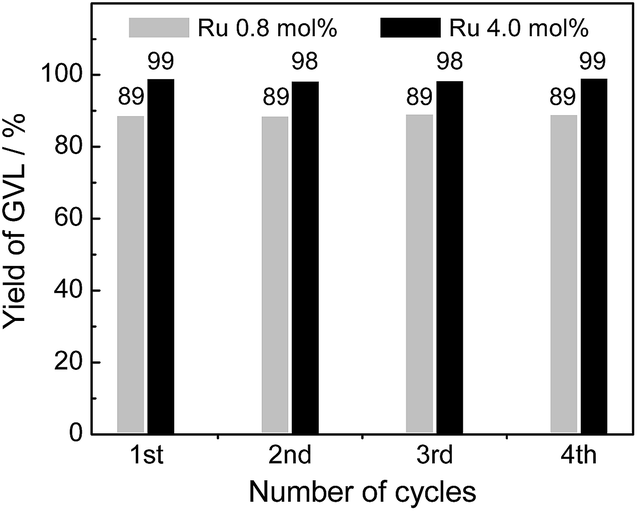 | ||
| Fig. 5 Recyclability test of 4% Ru(OH)x/TiO2(A) catalyst. Reaction conditions: catalyst (Ru 0.8 or 4.0 mol%), methyl levulinate (1 mmol), 2-PrOH (5 mL), 90 °C, 24 h, Ar. | ||
Experimental
Materials
All commercially available chemical reagents for material synthesis and catalytic tests were purchased from (Wako Pure Chemical Ind., Ltd.) and used without further purification. A variety of metal oxides were used as supports for Ru(OH)x catalyst; anatase TiO2 (ST-01, SBET = 339 m2 g−1) and rutile TiO2 (TTO-55, SBET = 47 m2 g−1) were supplied from Ishihara Sangyo Co., Ltd. Anatase and rutile TiO2 (P25, SBET = 55 m2 g−1) was supplied from AEROSIL Japan Industries. α-Al2O3 (AKP-3000, SBET = 10 m2 g−1) and γ-Al2O3 (JRC-ALO-4, SBET = 210 m2 g−1) were supplied from Sumitomo Chemical Co., Ltd. and the Catalysis Society of Japan, respectively. ZrO2 (RC-100, SBET = 112 m2 g−1) and CeO2 (SBET = 131 m2 g−1) were obtained from Daiichi Kigenso Kagaku Kogyo Co., Ltd. MgO (SBET = 45 m2 g−1) and Mg–Al hydrotalcite (SBET = 67 m2 g−1) were commercially supplied from Wako Pure Chemical Ind., Ltd.Catalyst preparation
Supported Ru(OH)x catalysts were prepared by precipitation method in aqueous RuCl3 solution according to a previously reported method.48 Typically, 2.0 g of metal oxide support pretreated at 550 °C for 3 h in air was mixed with an aqueous RuCl3 solution (8.3 mM) for 15 min with magnetic stirring. The solution pH was adjusted to 13.2 by addition of an aqueous NaOH solution (1 M), followed by stirring at room temperature for 24 h. The solid was then filtered, washed with a copious amount of water, and dried under vacuum to afford supported Ru(OH)x as a black powder. Pure Ru(OH)x was synthesized with a similar procedure except for the addition of oxide support.Characterization
Powder XRD patterns were recorded on a Rigaku RINT 2000 diffractometer with CuKα radiation (λ = 1.54056 Å, 40 kV to 40 mA). Nitrogen adsorption–desorption isotherms were measured at −196 °C by using Micromeritics ASAP 2020 system. The samples were degassed at 200 °C under vacuum for 4 h prior to the measurements to vaporize physisorbed water. The specific surface area was calculated by the BET (Brunauer–Emmett–Teller) method by using adsorption data ranging from P/P0 = 0.05 to 0.30. Elemental analysis was performed by Sumika Chemical Analysis Service. Ru loadings were determined by inductively coupled plasma atomic emission spectrometry (ICP-AES).Ru K-edge X-ray absorption spectra were recorded in fluorescence mode at room temperature at BL01B1 facility of SPring-8, Hyogo, Japan. All EXAFS data were examined by using REX2000 software (Rigaku), by which Fourier transformation of k3-weighted normalized EXAFS data (FT-EXAFS) was performed over the range 3.0 < k (Å−1) < 11.0 to obtain the radial structure function (RDF). For the curve-fitting analysis, the empirical phase shift and amplitude functions for Ru–O and Ru⋯Ru were extracted from the data for RuO2.
Procedures for catalytic reactions
In a typical reaction, catalyst (Ru 0.8 mol%), levulinate ester (1 mmol) and alcohol (5 mL) were added to a quartz glass reactor equipped with a reflux condenser and magnetically stirred at 90 °C under 1 atm of Ar. After the allotted reaction time, a portion of the reaction mixture was withdrawn by filtration and then analyzed by a gas chromatograph (Shimadzu GC-14B) with a flame ionization detector equipped with a capillary column (ULBON HR-20 M; 0.53 mm × 30 m; Shinwa Chemical Ind., Ltd.). Conversion of levulinate ester and yields of GVL and other products were determined by using biphenyl as an internal standard. For recycle test of catalyst, the spent catalyst was recovered by filtration, washed with acetone and water, dried under vacuum, and then subjected to next catalytic run.Conclusions
GVL, an important feedstock chemical, could be produced from biomass-derived levulinate esters and secondary alcohols as H-donors via a mild catalytic transfer hydrogenation process over supported Ru(OH)x catalysts. In particular, Ru(OH)x supported on high-surface-area, anatase TiO2 containing highly-dispersed ultrasmall Ru(OH)x nano-clusters was demonstrated to be highly active for CTH even under mild reaction conditions with only 0.8 mol% of the catalysts. A further faster GVL production rate was obtained by adding appropriate heterogeneous bases, such as MgO and hydrotalcite, affording an almost quantitative yield of GVL after 24 h of reaction. The catalyst was reusable over multiple cycles without loss of catalytic performances, making this material a potential candidate for efficient GVL production under ambient reaction conditions.Acknowledgements
This work was financially supported by the Research Institute for Innovation in Sustainable Chemistry in the Institute of Advanced Industrial Science and Technology (AIST). The Ru K-edge X-ray absorption experiments were performed at BL01B1 facility of SPring-8, Hyogo, Japan (Proposal no. 2013A1403). We gratefully acknowledge Dr Yu Horiuchi (Osaka Prefecture Univ., Japan) for his assistance with the XAFS measurements.Notes and references
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS PubMed.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516 RSC.
- J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584 RSC.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238 RSC.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85 RSC.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110 CrossRef CAS PubMed.
- J.-P. Lange, J. Z. Vestering and R. J. Haan, Chem. Commun., 2007, 3488 RSC.
- D. Wang, S. H. Hakim, D. Martin Alonso and J. A. Dumesic, Chem. Commun., 2013, 49, 7040 RSC.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510 CrossRef CAS PubMed.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249 CrossRef CAS PubMed.
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657 CrossRef CAS PubMed.
- P. P. T. Sah and S.-Y. Ma, J. Am. Chem. Soc., 1930, 52, 4880 CrossRef CAS.
- T. Runge and C. Zhang, Ind. Eng. Chem. Res., 2012, 51, 3265 CrossRef CAS.
- Z. Wu, S. Ge, C. Ren, M. Zhang, A. Yip and C. Xu, Green Chem., 2012, 14, 3336 RSC.
- J.-P. Lange, W. D. van de Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437 CrossRef CAS PubMed.
- X. Hu and C.-Z. Li, Green Chem., 2011, 13, 1676 RSC.
- X. Hu, Y. Song, L. Wu, M. Gholizadeh and C.-Z. Li, ACS Sustainable Chem. Eng., 2013, 1, 1593 CrossRef CAS.
- K. Tominaga, A. Mori, Y. Fukushima, S. Shimada and K. Sato, Green Chem., 2011, 13, 810 RSC.
- Y. Kuwahara, W. Kaburagi, K. Nemoto and T. Fujitani, Appl. Catal., A, 2014, 476, 186 CrossRef CAS PubMed.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338 CrossRef.
- L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529 CrossRef CAS PubMed.
- D. J. Braden, C. A. Henao, J. Heltzel, C. C. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755 RSC.
- M. G. Al-Shaal, W. R. H. Wright and R. Palkovits, Green Chem., 2012, 14, 1260 RSC.
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688 RSC.
- J. M. Nadgeri, N. Hiyoshi, A. Yamaguchi, O. Sato and M. Shirai, Appl. Catal., A, 2014, 470, 215 CrossRef CAS PubMed.
- P. P. Upare, J.-M. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J.-S. Chang, J. Ind. Eng. Chem., 2011, 17, 287 CrossRef CAS PubMed.
- K. Yan, C. Jarvis, T. Lafleur, Y. Qiao and X. Xie, RSC Adv., 2013, 3, 25865 RSC.
- K. Yan, T. Lafleur, G. Wu, J. Liao, C. Ceng and X. Xie, Appl. Catal., A, 2013, 468, 52 CrossRef CAS PubMed.
- J. Deng, Y. Wang, T. Pan, Q. Xu, Q.-X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163 CrossRef CAS PubMed.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064 RSC.
- K. Yan, J. Liao, X. Wu and X. Xie, RSC Adv., 2013, 3, 3853 RSC.
- R. S. Assary, L. A. Curtiss and J. A. Dumesic, ACS Catal., 2013, 3, 2694 CrossRef CAS.
- J. Zhang, S. Wu, B. Li and H. Zhang, ChemCatChem, 2012, 4, 1230 CrossRef CAS.
- M. Chia and J. A. Dumesic, Chem. Commun., 2011, 47, 12233 RSC.
- X. Tang, L. Hu, Y. Sun, G. Zhao, W. Hao and L. Lin, RSC Adv., 2013, 3, 10277 RSC.
- X. Tang, H. Chen, L. Hu, W. Hao, Y. Sun, X. Zeng, L. Lin and S. Liu, Appl. Catal., B, 2014, 147, 827 CrossRef CAS PubMed.
- L. Bui, H. Luo, W. R. Gunther and Y. Román-Leshkov, Angew. Chem., Int. Ed., 2013, 52, 8022 CrossRef CAS PubMed.
- J. Wang, S. Jaenicke and G.-K. Chuah, RSC Adv., 2014, 4, 13481 RSC.
- Z. Yang, Y.-B. Huang, Q.-X. Guo and Y. Fu, Chem. Commun., 2013, 49, 5328 RSC.
- J. W. Kim, T. Koike, M. Kotani, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2008, 14, 4104 CrossRef CAS PubMed.
- K. Yamaguchi, T. Koike, J. W. Kim, Y. Ogasawara and N. Mizuno, Chem.–Eur. J., 2008, 14, 11480 CrossRef CAS PubMed.
- M. Boronat, A. Corma and M. Renz, J. Phys. Chem. B, 2006, 110, 21168 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD data, CO2-TPD data, TEM image and XAFS analysis data of the materials, in situ FTIR spectra and reaction kinetics data. See DOI: 10.1039/c4ra08074b |
| This journal is © The Royal Society of Chemistry 2014 |