
The formation of HOCO in the coadsorption of water and carbon monoxide on Pt3Ni(111)
A. Politanoa and
G. Chiarello*ab
aUniversità degli Studi della Calabria, Dipartimento di Fisica, 87036 Rende, CS, Italy. E-mail: gennaro.chiarello@fis.unical.it; Fax: +39-0984-494401; Tel: +39-0984-496157
bConsorzio Nazionale Interuniversitario di Scienze Fisiche della Materia, via della Vasca Navale, 84, 00146 Roma, Italy
First published on 15th September 2014
Abstract
High-resolution electron energy loss spectroscopy has been used to study the coadsorption of water and carbon monoxide at the Pt3Ni(111) surface. The analysis of the vibrational spectrum indicates the reaction between coadsorbates (CO and OH groups of ice) with the formation of HOCO. Di-σ-trans-HOCO species are stabilized on the surface at up to 165 K. For higher temperature, HOCO desorbs from the surface.
1 Introduction
The chemical–electrical energy conversion is one of the aims of research on sustainable energy.1 The key chemical reaction in this process is the electrocatalytic reduction of oxygen at the cathode of fuel cells.2 Such reaction (oxygen reduction reaction, ORR) takes place on electrodes that are modified by OHads.3 Dispersed Pt nanoparticles on amorphous high-surface-area carbon are the most common electrocatalysts used to accelerate ORR.4,5 Commercial applications based on this renewable technology are limited by the high cost of the required Pt catalyst. Thus, large-scale diffusion could be possible only upon improving the catalyst performance to reduce the Pt loading.6–8 For example, a five-fold improvement of catalytic activity for the ORR is required for the commercial implementation of fuel cell technology in transportation.9In many works, Stamenkovic and Markovic have shown that high catalytic activity for the ORR can be achieved on Pt bimetallic alloys (Pt3M, M = Fe, Co, Ni, etc.), due to the altered electronic structure of the Pt outermost layer and hence reduced adsorption of oxygenated spectator species (e.g., OH).10–14 The Pt3Ni(111) surface has an unusual electronic structure (d-band center position) and arrangement of surface atoms in the near-surface region. Ni atoms are not present at the outermost layer of the surface, which is a Pt skin.14–17 However, Ni atoms in the subsurface region induce charge polarization at the surface and suppresses the surface-states of Pt3Ni(111).11 Moreover, subsurface Ni atoms induce a downshift of the d-band center of atoms of the Pt skin with implications for surface chemical reactivity via decreased activation energies for chemical reactions.16 For Pt3Ni(111),13 the catalytic enhancement obtained for ORR is ten times higher with respect pure Pt. However, the presence of CO as a contaminant is inevitable in Pt-based electrodes.18 Even in ultra-high vacuum conditions, both Pt(111) and Pt3Ni(111) are rapidly poisoned by carbon monoxide.19 Therefore, the comprehension of chemical reactions between adsorbed carbon monoxide and water on Pt3Ni(111) deserves a particular attention to understand chemical reactions at realistic electrocatalysts.
Furthermore, the water gas shift (WGS) reaction (H2O + CO → H2 + CO2) plays a crucial role in many industrial processes such as ammonia synthesis, the conversion of syngas and steam reforming of methanol. Despite the deep interest toward WGS in metal/oxides, Au20 and Pt,20 the comprehension of mechanisms controlling the reaction is still unsatisfactory. Even the possible presence of intermediates is debated. The WGS reaction is catalyzed by Pt at temperatures from 523 to 573 K.21 However, the comprehension of the chemical reactions for lower temperature, i.e. under controlled conditions in ultra-high vacuum, is essential to understand the intermediate states leading to the conversion of CO.
Jacob et al.22 have found that water adsorption on Pt(111) is partially dissociative in the monolayer regime and this is expected to occur also for the Pt skin of Pt3Ni(111). The presence of dissociated water may play crucial role in the chemistry of monolayer water on the Pt skin with implications in the catalytic properties of electrocatalysis.
Following the reaction mechanisms between CO and H2O,21,23,24 the reaction of OH groups with CO can produce formate (HCOO), carboxyl (HOCO), and/or carbonate (CO3) species as key intermediates before the final production of CO2 and H2. Although formates and carbonates have been detected in many experiments studying the water gas shift on metal/oxide catalysts,25 the HOCO intermediate is favored on gold surfaces.26
Vibrational spectroscopy is a powerful tool for identifying chemisorbed species at surfaces and, moreover, the species generated by surface reactions.27–29 High-resolution electron energy loss spectroscopy (HREELS) is particularly suitable for such aims for its high energy resolution and surface sensitivity.30
Herein, we report on a HREELS study on CO + H2O on Pt3Ni(111) at 100 K. The analysis of the vibrational spectrum indicates that HOCO intermediates form on this surface. The di-σ-trans-HOCO species can be stabilized on the surface up to 165 K.
2 Experimental section
The experimental apparatus consists in an ultra-high vacuum (UHV) chamber equipped with standard facilities for surface characterization. Pt3Ni(111), Pt(111), and Ni(111) samples were purchased from MaTecK GmbH. The substrate was cleaned by repeated cycles of ion sputtering and annealing at 1300 K. Surface cleanliness and order were checked using Auger electron spectroscopy (AES) and low-energy electron diffraction (LEED) measurements, respectively.All investigated surfaces, i.e. Pt3Ni(111), Pt(111) and Ni(111), showed an excellent LEED pattern with bright spots.
Carbon monoxide and water have been introduced into the vacuum chamber by means of leak valves.
HREELS experiments were performed by using an electron energy loss spectrometer (Delta 0.5, SPECS) with an impinging energy of 4 eV. The low value of the primary electron beam energy ensures of the absence of electron-induced dissociation or ionization of chemical species, which can occur only for primary energies in the keV range.31 HREEL spectra were taken in the specular direction (angle of incidence 55° with respect to the sample normal) and the energy resolution was set at 3–4 meV.
3 Results and discussion
3.1 CO adsorption
Fig. 1 shows the loss spectra acquired for CO-saturated Pt3Ni(111), Pt(111), Ni(111) samples at room temperature.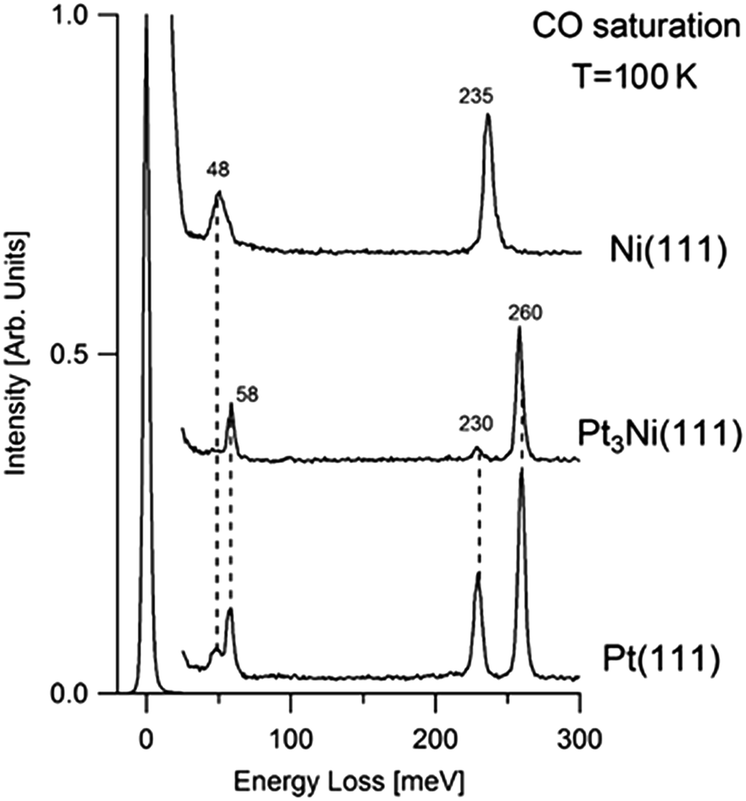 | ||
| Fig. 1 HREEL spectra for the CO-saturated Pt(111), Pt3Ni(111), and Ni(111) surfaces. All measurements and exposures have been carried out at 100 K. Spectra have been recorded in specular geometry (incidence angle 55° with respect to the sample normal) and with a primary electron beam energy of 4 eV. | ||
The same vibrational modes have been obtained for both Pt3Ni(111) and Pt(111). The loss features at 48 and 58 meV arise from the vibration of the whole CO molecule, adsorbed at bridge (mode at 48 meV) and atop sites (peak at 58 meV), against the Pt(111) substrate.32–34 Vibrational peaks at 230 and 258 meV arise from the intramolecular stretching vibration of CO adsorbed at bridge and atop sites, respectively, in agreement with previous results for CO adsorption on Pt(111)35–38 and other transition-metal systems.39 However, a preferential occupation of the atop site with respect to the bridge is observed for CO/Pt3Ni(111).
By contrast, the vibrational spectrum of CO-saturated Ni(111) is characterized by single CO–Ni and C–O stretching vibrations at 48 and 235 meV, respectively. The only adsorption site occupied by CO molecules is the three-fold hollow site.
One of the reasons leading to the improved ORR of Pt3M samples is their superior resistance to poisoning effect toward CO and OH in comparison to pure Pt.15 However, present results in UHV conditions demonstrate that both Pt3Ni(111) and Pt(111) surface strongly interact with CO molecules and, moreover, there are no noticeable differences between the two surfaces. Loss spectra of Fig. 1 also indicate that the first layer of Pt3Ni(111) is composed exclusively of Pt atom (Pt-skin), in agreement with results in ref. 13, 40 and 41. The possible modifications in the electronic structure of this Pt overlayer induced by Ni atoms located in the second layer do not affect CO adsorption. Density functional theory calculations showed that the interaction of adsorbates with Pt3Ni alloy is notably weakened with respect to the pure Pt sample.40,42 This should be a direct consequence of the downshift of the localised d-band center in the alloy.40 However, such finding is not confirmed by present vibrational measurements as concerns CO adsorption.
3.2 Water coadsorption with CO
Fig. 2a shows the HREEL spectrum of the CO-saturated Pt(111) surface as a function of water exposure at 100 K. The recorded vibrational bands may be divided into four major regions (see ref. 43 for a review): frustrated translations at 30 meV; frustrated rotations, i.e. librations at 100 meV; H2O deformations, i.e. the scissoring band centered around 200 meV; and O–H stretching modes around 400–460 meV. The significant width of the broad vibrational bands can be interpreted as due to a superposition of several vibration modes associated with different coordination.44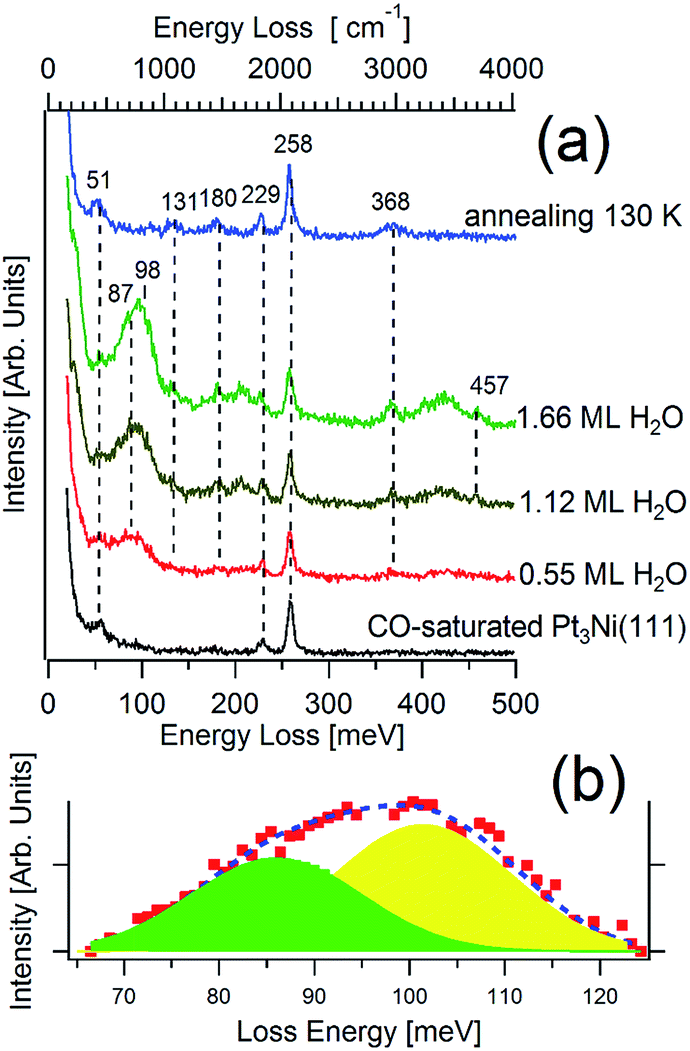 | ||
| Fig. 2 (a) HREELS spectrum of the CO-saturated Pt3Ni(111) (black curve), successively exposed to various amount of water at 100 K (red, brown, and green curve). Finally, the sample has been annealed at 130 K. (b) Fit with two Gaussian line-shapes of the librational band for 1.66 ML of water. The raw data have been fitted with a polynomial background. The primary energy is 4 eV. | ||
The centroid of the libration band abruptly shifts from 87 to 98 meV by increasing water coverage from 0.55 up to 1.66 ML (monolayer). The same effect has been observed for water on graphite and was interpreted as a fingerprint of a phase transition from a 2D to a 3D structure.44,45 Fig. 2b shows a fit of the librational band for 1.66 ML, showing that the intensity of the component at 102 meV is double with respect to that of the librational band at 87 meV. This situation is typical of ice coverage inferior than the bilayer. In particular, the librational mode at 102 meV, observed for the highest water coverage, represents the most intense librational vibration of ice I,44 in which water molecules are hydrogen-bonded to other four molecules. Water molecules with lower coordination have a corresponding lower energy of the librational mode.
The presence of a sharp O–H peak at 457 meV indicates the presence of OH groups which are not hydrogen-bonded.46 This relatively narrow transition is spectrally well separated from the much broader and red-shifted hydrogen-bonded part of the OH-stretch band. Therefore, the free OH-stretch mode at the interface represents a unique probe of the surface hydrogen bonds.47 Despite the O–H stretching band has been widely studied,48 the interpretation of spectral features, and in particular of the O–H stretching of free OH groups, is still highly controversial. Du et al.49 have assumed that the free-OH stretching mode is a fingerprint of hydrophobicity49 and, thus, it should be recorded only for hydrophobic surfaces. However, this mode has been observed also for hydrophilic substrates such as Pt(111), for both water clusters50 and bilayer ice.51 Instead, it has not been revealed in the HREEL spectrum of ice on graphite,44 which is known to be hydrophobic. Thus, the existence of H-free OH groups in CO + H2O/Pt3Ni(111) should just be interpreted as a boundary effect. Such conclusion is supported by recent findings by Ibach,52 who has reported that a band at 3690 cm−1 (457.5 meV) can be observed on H2O/Au(115) but not for water adsorbed on flat Au surfaces. This result has been explained by assuming that water adsorbed on the stepped surfaces possesses a considerable fraction of non-H-bonded OH groups whereas on the flat surfaces a complete saturation of H bonds occurs.
Dosing water onto the CO-modified Pt3Ni(111) does not produce a softening of the C–O stretching (νCO) for CO.
The overall lack of a change in water vibrational features in the CO + H2O phase suggests that on Pt(111) the coadsorbates separate into incompressible islands containing only water and compressible, internally repulsive, patches containing only CO. For CO + H2O/Pt(111), C–O bands associated with both atop and bridge sites considerably shift in the coadsorbed phase.53 Similar results have been obtained in the same coadsorption system on Rh(111).54
Thus, we can infer that water does not assemble into hydrogen-bonded islands microscopically intermixed with the contiguous CO adlayer regions.
Apart water- and CO-derived vibrational modes, the spectrum shows also additional features. The most striking result is the presence of a vibrational mode at 368 meV. Such frequency is typical of a C–H stretching vibration.55
Although CO does not dissociate on the clean Pt3Ni(111) surface, in principle the presence of water could induce partial CO dissociation at low temperatures. Likewise, CO dissociation has also been observed on Ir(111) in the presence of coadsorbed water.56 However, such mechanism is not effective at 100 K. The presence of a vibrational feature at 128 meV can be instead taken as fingerprint of HOCO formation. In fact, it should be ascribed to the out-of-plane H wagging mode.57 The peak at 365 meV is a normal mode of HOCO in which oxygen atoms are at rest, while C and H have a stretching motion.
Apart the CO symmetric stretch at 180 meV, the other vibrational features of HOCO (OCO bend) are overshadowed by the vibrational modes of the ice network.43,46,58
There are different adsorption structures of HOCO on Pt3Ni(111) surface (see Fig. 3). The most stable configuration is the di-σ-trans-HOCO, which could act as the precursor for the HOCO decomposition. HOCO intermediates are usually unstable at higher temperature but they are stabilized at 100 K. We would not be able to spectroscopically follow the successive HOCO decomposition since we can probe only adsorbed species, while both H2 and CO2 desorb from the surface and, moreover, such process is inhibited at 100 K. Thus, in the following we will focus on the formation of the HOCO intermediate.
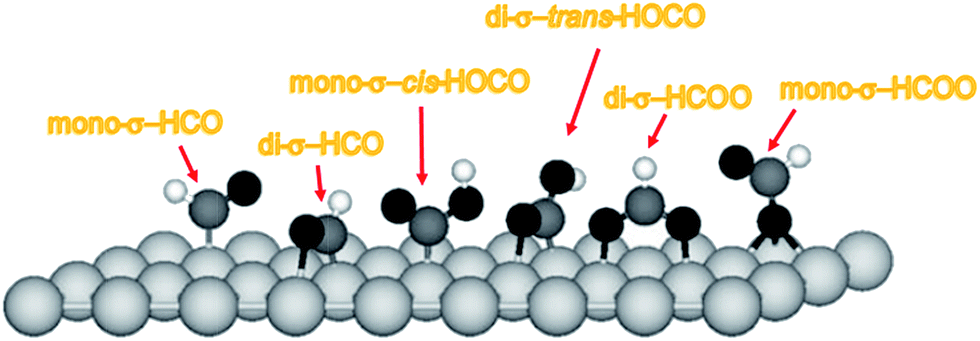 | ||
| Fig. 3 Sketch of the most stable mono-σ and di-σ adsorption configurations for HCO, HCOO and HOCO on Pt3Ni(111). Atomic spheres: black, O; white, H; dark gray, C; slight gray, Pt skin of the Pt3Ni(111) surface. | ||
There are two channels for the formation of the di-σ-trans-HOCO from CO and OH (Table 1). The first channel contains three elementary steps. The first step is CO + OH → mono-σ-cis-HOCO, whose barrier and reaction heat are reported in ref. 59 to be 41 and 15 kJ mol−1, respectively. The second one is the conversion of mono-σ-cis-HOCO to di-σ-cis-HOCO, with a barrier of only 2 kJ mol−1.59 The last step is the isomerization of the di-σ-cis- HOCO into the most stable di-σ-trans-HOCO, whose activation energy is 39 kJ mol−1.59 The second channel for the formation of the di-σ-trans-HOCO is the direct combination of CO and OH. The barrier for this channel is 49 kJ mol−1,59 that is only 8 kJ mol−1 higher than that of the first step of the first channel. Thus, the second channel also contributes to the formation of di-σ-trans-HOCO.59
| Elementary reactions | Activation energy [kJ mol−1] |
|---|---|
| C1.1: CO + OH → mono-σ-cis-HOCO | 41 |
| C1.2.: Mono-σ-cis-HOCO → di-σ-cis-HOCO | 2 |
| C1.3: Di-σ-cis-HOCO → di-σ-trans-HOCO | 39 |
| C2: CO + OH → di-σ-trans-HOCO | 49 |
We cannot exclude the formation at the surface of formate (HCOO) together with carboxylic (HOCO) species. However, the adsorption energy of formate species is just one half with respect to that of carboxylic groups59 and thus such reaction is less favored even if possible. Therefore, the [HOCO]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[HCOO] stoichiometry will be high.
:[HCOO] stoichiometry will be high.
After heating at 130 K (blue spectrum in Fig. 2a), ice desorbs while features arising from CO and HOCO remain in the spectrum.
After saturating with CO 2.5 ML of water, a higher content of HOCO on the surface can be inferred from the analysis of Fig. 4. Upon annealing at 165 K, the vibrational spectrum shows only features coming from molecular CO. Thus, HOCO species are not stable at 165 K. We also note that non-H bonded OH groups are nearly absent after the completion of the bilayer, as indicated by the lack of the free-OH O–H stretching at 457 meV in the vibrational spectrum for 2.5 ML (red curve in Fig. 4).
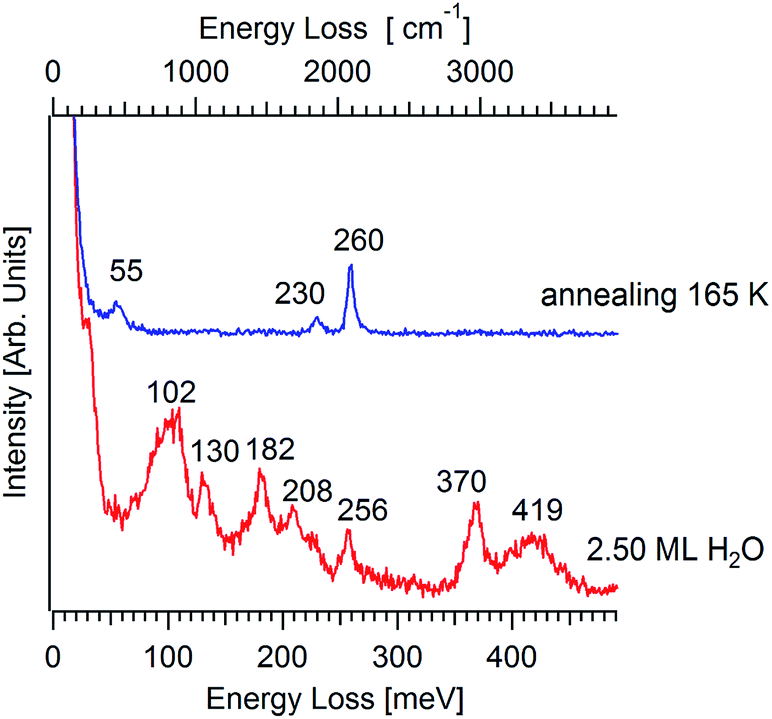 | ||
| Fig. 4 HREELS spectrum for 2.5 ML of H2O (red curve) on Pt3Ni(111). Successively, the sample has been heated at 165 K (blue curve). The primary energy is 4 eV. | ||
In Fig. 5a we report on the competitive adsorption of water and carbon monoxide. In correspondence of carbon monoxide saturation, a c(4 × 2) LEED pattern is observed, which corresponds to 0.5 ML. In a CO-saturated surface, water does not adsorb. By decreasing CO coverage, water adsorption starts to occur. The HREELS signal derived by HOCO reaches a maximum for a ratio between the [H2O]/[CO] coverages of 0.1 (Fig. 5b) while for higher values of [H2O]/[CO] a decreasing behavior of the vibrational intensity of HOCO is found.
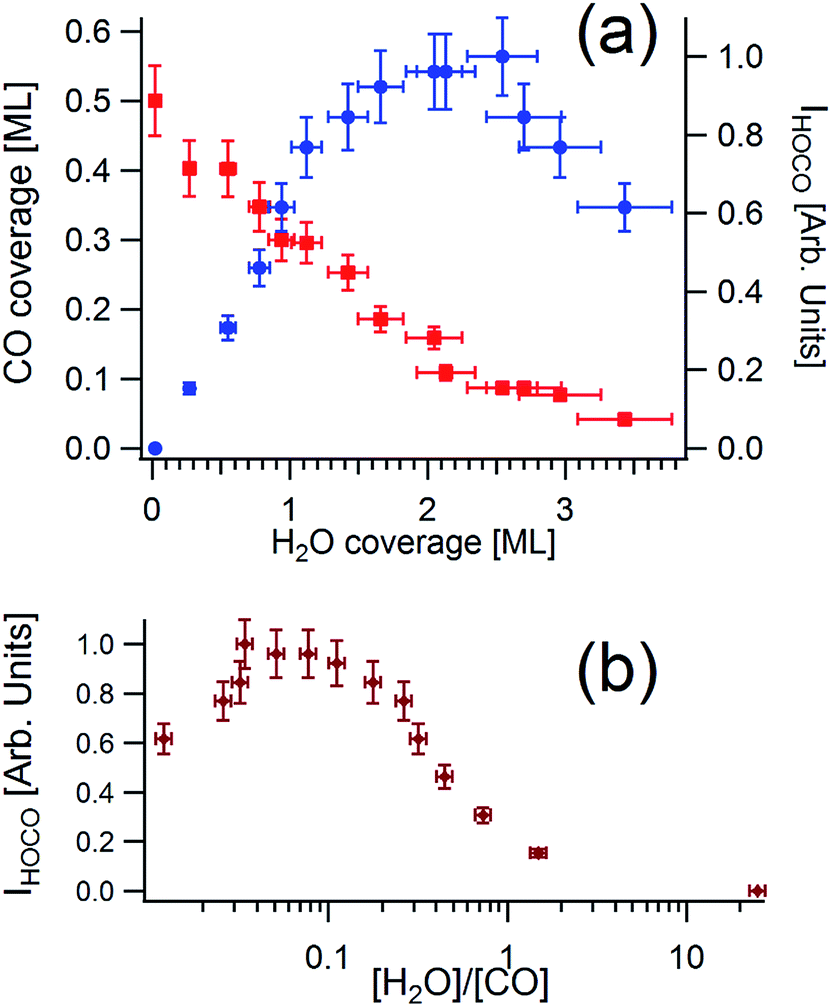 | ||
| Fig. 5 (a) CO coverage (red squares) versus H2O coverage at 100 K. Blue circles indicate the intensity of the vibrational feature at 368–370 meV, fingerprint of HOCO formation. (b) Intensity of the HOCO mode at 368–370 meV, plotted as a function of the ratio between H2O and CO coverage. | ||
4 Conclusions
We have studied the coadsorption of carbon monoxide and water on Pt3Ni(111). We find that di-σ-trans-HOCO species can be stabilized at surface in a temperature range up to 165 K. Thus, we can infer that the WGS reaction on Pt3Ni proceeds with HOCO intermediates. This work claims for addressed theoretical works aimed at understanding the WGS reaction at Pt3M catalysts.Acknowledgements
AP thanks Regione Calabria for salary support.References
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS PubMed.
- S. Mukerjee and S. Srinivasan, J. Electroanal. Chem., 1993, 357, 201–224 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Z. Liu, L. M. Gan, L. Hong, W. Chen and J. Y. Lee, J. Power Sources, 2005, 139, 73–78 CrossRef CAS PubMed.
- Z. Wen, J. Liu and J. Li, Adv. Mater., 2008, 20, 743–747 CrossRef CAS.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781–787 CrossRef CAS PubMed.
- J.-H. Wee, K.-Y. Lee and S. H. Kim, J. Power Sources, 2007, 165, 667–677 CrossRef CAS PubMed.
- V. Neburchilov, H. Wang and J. Zhang, Electrochem. Commun., 2007, 9, 1788–1792 CrossRef CAS PubMed.
- S. J. C. Cleghorn, X. Ren, T. E. Springer, M. S. Wilson, C. Zawodzinski, T. A. Zawodzinski and S. Gottesfeld, Int. J. Hydrogen Energy, 1997, 22, 1137–1144 CrossRef CAS.
- B. Fowler, C. A. Lucas, A. Omer, G. Wang, V. R. Stamenković and N. M. Marković, Electrochim. Acta, 2008, 53, 6076–6080 CrossRef CAS PubMed.
- Y. S. Kim, S. H. Jeon, A. Bostwick, E. Rotenberg, P. N. Ross, V. R. Stamenkovic, N. M. Markovic, T. W. Noh, S. Han and B. S. Mun, Adv. Energy Mater., 2013, 3, 1257–1261 CrossRef CAS.
- V. Stamenković, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Marković, J. Rossmeisl, J. Greeley and J. K. Nųrskov, Angew. Chem., 2006, 45, 2897–2901 CrossRef PubMed.
- V. R. Stamenković, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef PubMed.
- D. F. van der Vliet, C. Wang, D. Li, A. P. Paulikas, J. Greeley, R. B. Rankin, D. Strmcnik, D. Tripkovic, N. M. Markovic and V. R. Stamenkovic, Angew. Chem., 2012, 124, 3193–3196 CrossRef.
- V. R. Stamenković, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Marković, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef PubMed.
- H.-Y. Su, X.-H. Bao and W.-X. Li, J. Chem. Phys., 2008, 128, 194707 CrossRef PubMed.
- Z. Yang, J. Wang and X. Yu, Chem. Phys. Lett., 2010, 499, 83–88 CrossRef CAS PubMed.
- X. Cheng, Z. Shi, N. Glass, L. Zhang, J. Zhang, D. Song, Z.-S. Liu, H. Wang and J. Shen, J. Power Sources, 2007, 165, 739–756 CrossRef CAS PubMed.
- G. Chiarello, A. R. Marino, V. Formoso and A. Politano, J. Chem. Phys., 2011, 134, 224705 CrossRef CAS PubMed.
- M. Gonzalez Castaño, T. R. Reina, S. Ivanova, M. A. Centeno and J. A. Odriozola, J. Catal., 2014, 314, 1–9 CrossRef PubMed.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, J. Phys. Chem. C, 2008, 112, 4608–4617 CAS.
- T. Jacob and W. A. Goddard, J. Am. Chem. Soc., 2004, 126, 9360–9368 CrossRef CAS PubMed.
- R. A. Ojifinni, N. S. Froemming, J. Gong, M. Pan, T. S. Kim, J. M. White, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 6801–6812 CrossRef CAS PubMed.
- A. A. Gokhale, J. A. Dumesic and M. Mavrikakis, J. Am. Chem. Soc., 2008, 130, 1402–1414 CrossRef CAS PubMed.
- R. Burch, Phys. Chem. Chem. Phys., 2006, 8, 5483–5500 RSC.
- P. Liu and J. A. Rodriguez, J. Chem. Phys., 2007, 126, 164705 CrossRef PubMed.
- A. Politano and G. Chiarello, J. Phys. Chem. C, 2011, 115, 13541–13553 CAS.
- A. Politano, G. Chiarello, G. Benedek, E. V. Chulkov and P. M. Echenique, Surf. Sci. Rep., 2013, 68, 305–389 CrossRef CAS PubMed.
- G. Chiarello, A. Cupolillo, C. Giallombardo, R. G. Agostino, V. Formoso, D. Pacilé, L. Papagno and E. Colavita, Surf. Sci., 2003, 536, 33–44 CrossRef CAS.
- H. Ibach and D. L. Mills, Electron Energy Loss Spectroscopy and Surface Vibrations, Academic Press, San Francisco, 1982 Search PubMed.
- F. Alvarado, R. Hoekstra and T. Schlathölter, J. Phys. B: At., Mol. Opt. Phys., 2005, 38, 4085 CrossRef CAS.
- N. R. Avery, J. Chem. Phys., 1981, 74, 4202–4203 CrossRef CAS PubMed.
- A. M. Baro and H. Ibach, J. Chem. Phys., 1979, 71, 4812–4816 CrossRef CAS PubMed.
- S. Ohnishi and N. Watari, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14619 CrossRef CAS.
- X. Su, P. S. Cremer, Y. R. Shen and G. A. Somorjai, Phys. Rev. Lett., 1996, 77, 3858 CrossRef CAS.
- W. D. Mieher, L. J. Whitman and W. Ho, J. Chem. Phys., 1989, 91, 3228–3239 CrossRef CAS PubMed.
- C. Klünker, M. Balden, S. Lehwald and W. Daum, Surf. Sci., 1996, 360, 104–111 CrossRef.
- S. Moon-Bong, Y. Koichiro and I. Masatoki, Chem. Phys. Lett., 1996, 263, 585–590 CrossRef.
- A. Politano, V. Formoso and G. Chiarello, J. Phys. Chem. C, 2009, 113, 316–320 CAS.
- D. Pillay and M. D. Johannes, J. Phys. Chem. C, 2008, 112, 1544–1551 CAS.
- V. Stamenković, T. J. Schmidt, P. N. Ross and N. M. Marković, J. Electroanal. Chem., 2003, 554–555, 191–199 CrossRef.
- D. Pillay and M. D. Johannes, Surf. Sci., 2008, 602, 2752–2757 CrossRef CAS PubMed.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
- D. V. Chakarov, L. Österlund and B. Kasemo, Vacuum, 1995, 46, 1109–1112 CrossRef CAS.
- D. V. Chakarov, L. Österlund and B. Kasemo, Langmuir, 1995, 11, 1201–1214 CrossRef CAS.
- A. Politano and G. Chiarello, J. Chem. Phys., 2013, 139, 064704 CrossRef PubMed.
- P. N. Perera, K. R. Fega, C. Lawrence, E. J. Sundstrom, J. Tomlinson-Phillips and D. Ben-Amotz, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12230–12234 CrossRef CAS PubMed.
- S. Meng, L. F. Xu, E. G. Wang and S. Gao, Phys. Rev. Lett., 2002, 89, 176104 CrossRef.
- Q. Du, E. Freysz and Y. R. Shen, Science, 1994, 264, 826–828 CAS.
- M. Nakamura, Y. Shingaya and M. Ito, Chem. Phys. Lett., 1999, 309, 123–128 CrossRef CAS.
- S. Meng, Surf. Sci., 2005, 575, 300–306 CrossRef CAS PubMed.
- H. Ibach, Surf. Sci., 2012, 606, 1534–1541 CrossRef CAS PubMed.
- N. Kizhakevariam, X. Jiang and M. J. Weaver, J. Chem. Phys., 1994, 100, 6750–6764 CrossRef CAS PubMed.
- F. T. Wagner, T. E. Moylan and S. J. Schmieg, Surf. Sci., 1988, 195, 403–428 CrossRef CAS.
- A. Politano and G. Chiarello, Carbon, 2013, 61, 412–417 CrossRef CAS PubMed.
- M. Pan, S. Hoang, J. Gong and C. B. Mullins, Chem. Commun., 2009, 7300–7302 RSC.
- D. A. Dixon, D. Feller and J. S. Francisco, J. Phys. Chem. A, 2002, 107, 186–190 CrossRef.
- A. Politano, A. R. Marino, V. Formoso and G. Chiarello, Carbon, 2011, 49, 5180–5184 CrossRef CAS PubMed.
- Q.-L. Tang, Z.-X. Chen and X. He, Surf. Sci., 2009, 603, 2138–2144 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |