
Modulating the polyoxometalate-based inorganic–organic hybrids from simple chains to complicated frameworks via changing POM clusters†
Abstract
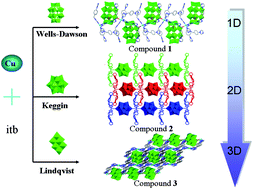
Investigation into a hydrothermal reaction system with a copper salt, 1-(imidazo-1-ly)-4-(1,2,4-triazol-1-ylmethyl)benzene) (itb) ligand and three types of polyoxometalates (POMs) led to the preparation of three new coordination polymers, [Cu2(itb)4(H4P2W18O62)]·H2O (1), [Cu(itb)4(HPW12O40)]·4H2O (2) and [Cu2(itb)2(Mo8O26)]·6H2O (3). The three coordination polymers were characterized by elemental analyses, single-crystal X-ray diffraction, powder X-ray diffraction, XPS and IR spectra. Structural analyses show that with modulating the type of POM anions from [Mo8O26]4− and [PW12O40]3− to [P2W18O62]6−, the three coordination polymers possess distinct structural motifs: simple one-dimensional (1D) chain (1), 1D + 1D → 2D interdigitated architectures (2) and complicated 3D frameworks consisting of both meso-helixes and left/right-helixes (3). The distinct structural features of the three coordination polymers suggest that the POM anions should play a significant role in the process of assembly. The electrocatalytic and luminescent properties for coordination polymers 1–3 have been investigated.
 Please wait while we load your content...
Please wait while we load your content...

